
最新MISEV外泌體國際標準指南
台灣諾曼達科技為TSEV(台灣胞外體學會)指定合作夥伴, 提供國際權威認證的完整平台和最專業的相關知識, 在外泌體(Exosome) / 細胞外囊泡(Extracellular vesicles)相關之研究、製造、品管等方面皆已有多篇著名期刊引用, 目前已在國內各單位舉辦超過30場外泌體研究與製程分析講座, 如有相關需求歡迎聯絡洽詢。
做為在外泌體領域的指定合作單位, 我們替您解析了MISEV2018的內容如下:
 |
-
-
命名法
細胞外囊泡(Extracellular Vesicles, EVs)指的是經細胞釋放而具有脂質雙分子層的顆粒, 不能複製即不包含具有功能的核心
而外泌體(Exosomes)和微囊泡(Microvesicles)都是EVs的亞分類, 目前並沒有單一技術可完全證明所屬是哪一種, 這代表我們需要建立一個完整的驗證模式, 比如依據EVs的物理特性、生化成分、處理條件、細胞起源或是依自有的發現法命名, 如果無法依照 MISEV2018 的方法命名, 我們的建議是用微粒(Extracellualr Particles, EP)取代
可是外泌體(Exosomes)的說法實在很吸引人, 相較下EVs的稱呼似乎沒這麼好用, 我們可以建議您針對計劃書、專業性期刊或是囊泡相關的期刊的說法, 歡迎進一步聯絡我們-
樣品的前處理
包含細胞條件培養液、生物體液和組織等類型
如何去除細胞條件培養液中FBS的EVs -
1. 配置好的培養液(或 20% FBS 培養液), 以 100,000g/18 hrs 離心
2. 配置好的培養液(或 20% FBS 培養液), 以 200,000g/1 hr 離心
3. 使用切向流過濾或是超濾法 (100Kd)
以上的建議還是有補充的注意事項而不一定都可接受, 歡迎進一步聯絡我們-
樣品的儲存
目前已知的保存條件都還可接受並沒有通用的共識形成, 雖然不會影響顆粒濃度但會有粒徑分佈的變化(聚集), 在功能研究的影響不大
-
分離方法的選擇和純度評估
依 MISEV2018 的說法, 提取的方法可分為:
1. 高回收, 低純度 : 主要是目前市面上各種以 PEG 為主的沉澱試劑盒 (Kit)、低分子量截流的超濾、UC 超速離心法等
2. 適中回收, 適中純度 : 包含 SEC 尺寸排阻色譜法、高分子量截流的超濾、差速離心法、切向流過濾、親合層析法
3. 低回收, 高純度 : 結合以上的兩種方式, 例如 UC + SEC, 密度梯度離心或免疫親和捕獲
4. 高回收, 高純度 : 目前還沒有已被承認的方式
那麼要如何選擇合適的提取方法呢, 主要還是依照您的實驗目標和樣品種類來考量, 歡迎進一步聯絡我們-
細胞外囊泡標記物的表徵策略
依 MISEV2018 的說法, 要有兩個陽性蛋白和一個陰性蛋白標記物, 我們的建議是先做跨膜蛋白 Tetraspanins(主要是 CD63, CD8, CD81, CD82)再做膜內標記(主要是 Allx, Tsg101)
在 MISEV2018 Table 3. 內有基於蛋白質含量的 EV 表徵描述 -
1. 與質膜和/或內體有關的跨膜或GPI錨定蛋白
2. EVs 中保留的胞漿蛋白
3. 非 EVs 共分離結構的主要組成部分
4. 除 PM/內體外與其他細胞內區室相關的跨膜、脂質結合和可溶性蛋白
5. EVs 保留的分泌蛋白
第4點和第5點一般為有特異來源時才考慮, 歡迎進一步聯絡我們對於單一囊泡的表徵, 需要使用兩種不同但互補的方法:
1. 例如 AFM 的 SPM 和高分辨率的顯微鏡, 注意要同時提供專一的圖像和廣泛的圖像
2. 利用顆粒分析技術, 例如 TRPS、NTA、FC 等-
EVs 的定量和樣品歸一化
對顆粒濃度(數目): TRPS、NTA、FACS、AFM
對蛋白定量: Bradford / BCA(總蛋白)、ELISA(特定蛋白)
對脂質或RNA總量: 目前尚無已成熟的策略, 仍在確定中
對於歸一化, 目前EVs研究中還沒有公定的內參, 但文獻內有使用外參的說法, 歡迎進一步聯絡我們-
確認特定分子與EVs的拓樸結構
主要是確定所屬分子是在囊泡上或是囊泡內, 抑或是在提取時共沉澱形成的; 注意小心的設置對照組, 歡迎進一步聯絡我們
例如:
1. 未經過任何處理的樣品
2. 只用降解酶處理的樣品
3. 使用降解酶和洗滌劑處理
4. 只用洗滌劑處理-
證明EVs及其內容物與功能的關聯
法1: 以密度梯度提取EVs, 在功能研究中分別測定每個梯度的活性表現
法2: 以SEC管柱提取EVs, 在功能研究中分別測定每個餾份的活性表現
法3: 比較完整的EVs和經洗滌劑處理的活性, 但此法會有些技術的侷限性, 歡迎進一步聯絡我們
法4: 以過量的免疫親和捕獲以去除實驗內的EVs, 檢測此和對照組的功能是否有對應現象
總之, 原理即是去除EVs中的特定分子, 確認功能研究中的EVs是否不再具有對應現象, 也有使用電轉去除法(Electroporation)以上內容即非經許可請勿任意複製轉發分享, 如有任何建議或詢問, 歡迎和我們聯繫
-
台灣諾曼達科技為TSEV(台灣胞外體學會)指定合作夥伴, 提供國際權威認證的完整平台和最專業的相關知識, 在外泌體(Exosome) / 細胞外囊泡(Extracellular vesicles)相關之研究、製造、品管等方面皆已有多篇著名期刊引用, 目前已在國內各單位舉辦超過30場外泌體研究與製程分析講座, 如有相關需求歡迎聯絡洽詢。
做為在外泌體領域的指定合作單位, 我們替您解析了 MISEV2023的內容如下:
 |
摘要
細胞外囊泡(EVs)通過其複雜的貨物, 可以反映其來源細胞的狀態並改變其他細胞的功能和表型。
這些特徵表明了強大的生物標誌物和治療潛力, 並引起了廣泛的興趣, 有關EVs的科學出版物數量逐年穩定增長就是證明。
然而,由於在EV的命名、非囊泡狀細胞外顆粒的分離、表徵和功能研究等方面的挑戰, 要想實現EVs在從基礎生物學到臨床應用等領域的潛力仍存在障礙。
為了應對這一快速發展領域的挑戰和機遇, 國際細胞外囊泡學會(ISEV)更新了其”細胞外囊泡研究的最低資訊”, 該資訊於2014年首次發表, 隨後於2018年分別命名為MISEV2014和MISEV2018。
當前MISEV2023文件的目標是向研究人員提供可用方法的最新快照及其在從多種來源(包括細胞培養物、體液和固體組織)生產、分離和表徵EVs方面的優勢和局限性。
除了介紹EVs研究基本原理的最新技術水準之外, 本文件還涵蓋了目前正在拓展該領域邊界的先進技術和方法。
MISEV2023還包括關於EV釋放和攝取的新章節, 以及對研究EVs於體內方法的簡要討論。
本文彙集了來自ISEV專家工作組和1000多名研究人員的反饋, 傳達了EVs研究的現狀以促進可靠的科學發現, 並能更快地推進該領域。
以上內容即非經許可請勿任意複製轉發分享, 如有任何建議或詢問, 歡迎和我們聯繫
1. ISEV和MISEV的簡介
1.1 細胞外囊泡與ISEV
細胞外囊泡(EVs)在大多數生物系統中扮演著不同而重要的角色, 部分原因在於其組成的複雜性。
EVs是脂質雙層膜界定為奈米至微米大小的顆粒, 似乎可由所有的細胞類型釋放。EVs的分子和結構異質性意味著在基礎生物學以及生物標記物和治療應用的開發方面仍有許多發現要做, 但同樣的複雜性也給EVs研究的每個階段帶來了挑戰。
從定義和分類到分離、表徵、工程與臨床應用, ”細胞外囊泡研究的最低資訊” (MISEV) 旨在幫助EV研究和應用的所有相關從業人員以遵循每個特定問題和指示的最佳實踐。
現在是第三次迭代, MISEV2023做為一份現場共識文件, 旨在前兩次迭代中規定的標準和指南中, 為EV的相關研究提供建議和指導並鼓勵加強研究設計和實驗細節報告, MISEV是由國際細胞外囊泡協會(ISEV)所製作的(https://www.isev.org)。
ISEV成立於2011年其旨在加強全球的EV研究, 是參與細胞外囊泡相關領域科學家和臨床醫生的領先專業協會。
ISEV通過其年會、專題研討會和其他會議(面對面和虛擬)、同行評審期刊、線上學習平台以及與其他協會的夥伴關係, 吸引了世界各地不同的研究人員群體。
因此, ISEV處於獨特的地位並能夠引導關於最佳實踐指南和科學考慮相關專家共識的發展和傳播。
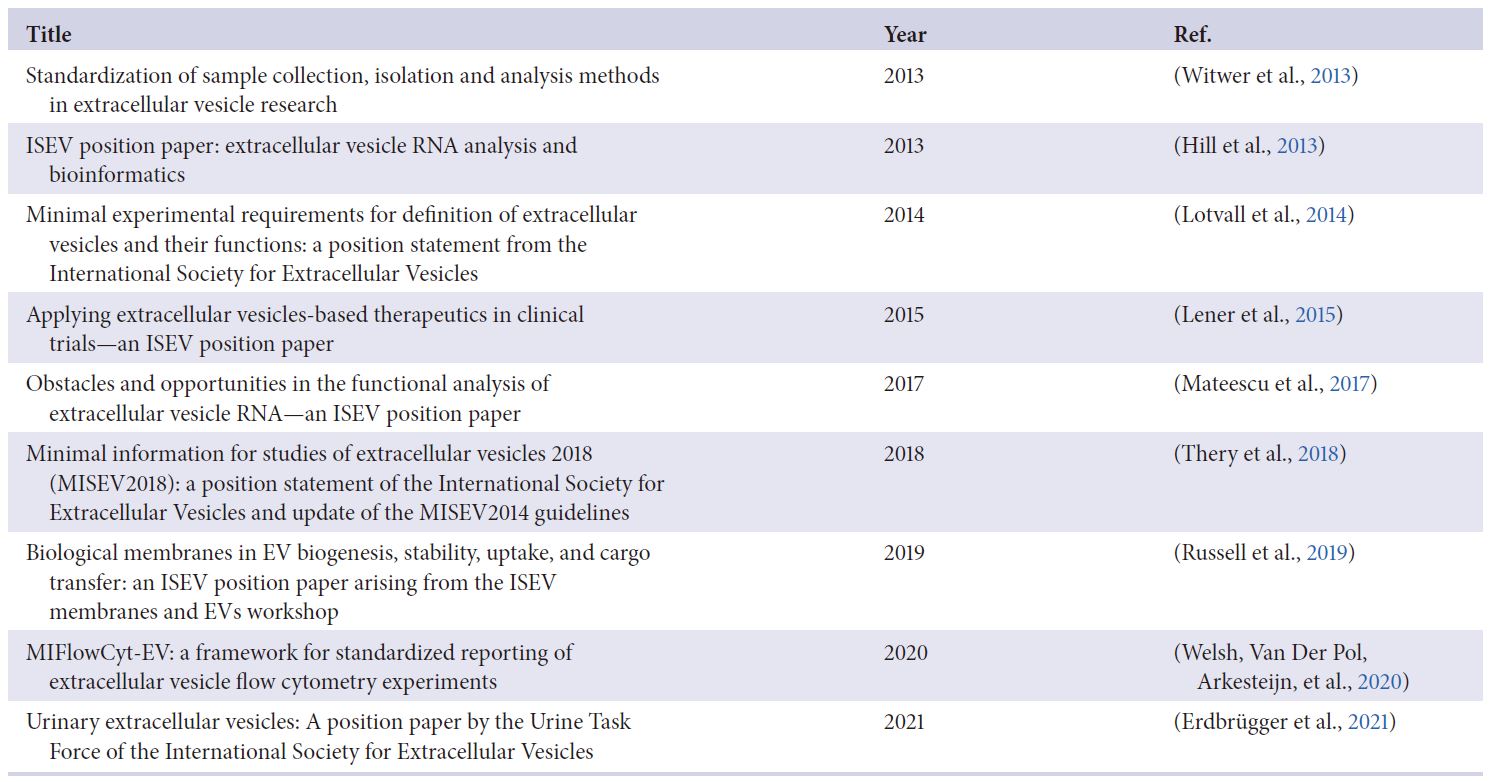
表1. JEV期刊: ISEV立場意見書與陳述
MISEV2014 (Lotvall等人發表, 2014年)是ISEV生產的第一份EV定位報告, 旨在為EV分析提供穩健性。
MISEV2018 (Thery等人發表, 2018)對推進該領域所採用的方法和手段進行了更深入和批判性的評估, 其中許多方法和手段至今仍然有效。MISEV2018還包括建議的實驗方法, 以應對一些剩餘挑戰並提供穩健的EV表徵。
早期MISEV的建議在很大程度上或許完全有效, 且應結合先前的文件來閱讀MISEV2023。
與之前的迭代一樣, MISEV2023為EV研究人員提供了簡明的建議和指導, 對MISEV2018中提出的問題進行了細化, 並為較新的開發領域增加了建議和指導, MISEV2023廣泛涵蓋了EVs的命名、預處理變數、分離與表徵以及EVs的釋放、攝取和功能性的體外和體內分析。
除了之前的MISEV指南之外, ISEV還促進並協調了包括學會內部立場意見書的相關最佳實踐指南和科學考慮的專家共識(Welsh, Van Der Pol, Arkesteijn等人, 2020), 且聚焦在特定主題的專家建議(Erdbrügger 等人發表, 2021; Hill 等人發表, 2013; Lener 等人發表, 2015; Mateescu 等人發表, 2017; Russell 等人發表, 2019; Verweij 等人發表, 2021; Witwer 等人發表, 2013) – 如表1.
最近, ISEV嚴謹性與標準化小組委員會監督特殊利益集團在EVs特定來源和其他EVs相關主題上的任命和活動, ISEV還建議採用其他的報告和圖集工具, 例如”用於即時逆轉錄酶定量聚合酶鏈式反應(qPCR)分析的定量即時PCR實驗出版最低資訊”(MIQE) (Bustin 等人發表, 2009)與EV-TRACK。
總體而言, ISEV的活動和建議都旨在提高EV研究的嚴密性、再現性和透明度。
本MISEV文件的目標是幫助所有在EV研究與應用的所有領域的從業人員在每個單獨的EV來源、類型、研究問題或應用之實施與開發。
1.2 MISEV是什麼? 並不是什麼?
自MISEV2018出現以來, 關於指南的含義以及該如何應用或不該如何應用已經進行了大量討論。根據討論情況, 現將MISEV的現狀和不現狀總結如下。
MISEV是:
- EVs研究簡介。
- 旨在提高EV研究設計、執行和報告期間的嚴密性、再現性和透明度的一組建議。
- 是一種工具, 可幫助審閱者和編輯利用其自身的專家知識, 評估與EV相關的提案、資助申請、摘要和手稿的優缺點。
- 各種有用的EVs技術和平台的非詳盡示例集。
- 一個支援創新型的EVs研究和應用以及從產品開發商到監管機構等各方的嚴格和標準化框架。
- 表明了當前在EVs領域以及一些不確定和增長領域的廣泛共識。
- 與轉化和臨床研究及應用相關, 包括治療性EVs的生產和初步評估。
- 適用於各種EVs研究和應用不僅僅是關於哺乳動物的來源。
儘管MISEV中提供的示例可能特定於哺乳動物EVs, 但基本原則很可能適用於所有EV來源, 其中包括資訊性術語、來源定義、分離/濃縮技術描述、EVs特性、適當控制的功能研究和綜合報告。
相比之下, MISEV並不是:
- 一個放之四海而皆準的標準, 一份全面的“該做和不該做”的清單或代替詳細的專家判斷; MISEV沒有絕對要求或禁止的技術或平台。同樣的MISEV也不強制使用任何特定的富集標記物, 所選擇的技術和目標應適合其研究目的與實驗系統而有助於總體MISEV的符合性。重要的是, 沒有一個研究團隊能夠接觸到所有的技術和平台。
- 創新的障礙。當引入EVs的新技術或新應用時, 該方法的某些方面可能不完全符合現有的MISEV框架, 或者更有可能不符合審查員對該框架的解釋。參見上文關於絕對授權的內容, 如有必要可援引例外條款; MISEV不應扼殺創新而應告知創新或新技術是如何展示和驗證的。
- 阻止出版或資助某一特定專案的手段。正如MISEV不應該扼殺創新一樣, 其也不應該被用來阻止與學會共用的研究成果; 例如: 不能證明生物發生的“外來體”或“外來體”研究可以改為關於EVs或“EV”報告, 但不能完全定性為更廣泛的細胞外顆粒研究, 這可能需要適當的控制來證明EVs的作用, 但如果無法做到, 則只需承認這些警告即可。
- 全面的引文集。每一篇都充分體現了MISEV的建議, MISEV文獻不是綜述或概要, 此處僅引用了一小部分的EV文獻, 且每一次引用都是出於特定目的。MISEV中的引用並不意味著ISEV認可某個報告、作者團隊、期刊或出版商, 也不表明所引用研究的首要性或完整性, 一些引用的研究也可能包含與MISEV建議不一致的方面; 此外,許多優秀的研究也沒有在本檔中引用。
總之, MISEV的精神體現在幾個問題中:
- 您使用什麼術語, 各別是什麼意思?
- 您從哪裡獲得EVs的?
- 您是如何分離、濃縮、表徵和儲存EVs的?
- 與其他成分相比, 您對EVs的功能或生物標誌物的歸屬有多少信心?
- 您是否充分詳細的共用了資料和報告方法, 以使其他人能夠複製或再現您的結果?
1.3 如何使用MISEV
- MISEV2023旨在幫助任何和所有的EVs研究人員:從剛開始EVs的新手到希望了解EVs行業當前的技術水準和(或)面臨到尖端問題的老牌研究人員。不過其結果是一個大文檔, 這可能會需要一些指導説明。
- 命名(第2章節)適用於所有的EV研究; 清晰且一致的語言將有助於確保結果是可理解和可比較的。
- 對於那些對EVs研究的新手, 我們建議第3–5章節非常重要, 分別涵蓋了樣本採集與處理、EV分離方法和EV表徵的最低考慮事項。
- 第6~第9章節為EV表徵、調節EV釋放和攝取的方法、EV功能研究和體內EV分析提供進一步的技術性指導, 這些章節為讀者提供了決策的最新資訊, 但大部分並沒有給出具體建議。
- 因此MISEV2023中提供的資訊和指南促進了EV科學的嚴謹性、再現性和透明度, 目的是確保所執行的實驗和報告的資訊可支援其結論。
共識: 89.3% (891)的MISEV2023調查受訪者“完全同意”, 10.7% (107)的受訪者“大部分同意”第1部分: ISEV和MISEV簡介; 沒有受訪者不同意(“大部分”或“完全”), 也沒有受訪者表示他們沒有意見和(或)專業知識。
以上內容即非經許可請勿任意複製轉發分享, 如有任何建議或詢問, 歡迎和我們聯繫
2. 術語 - EV定義和EV子分類
2.1 EV定義和EV子分類
定義: ”細胞外囊泡(EVs)”一詞是指從細胞中釋放出來的、由脂雙層界定且不能自行複製 (即不含功能性核) 的顆粒。
保留了MISEV2018中的當前EV定義,但刪除了2018年使用的”自然”(如: 自然釋放)一詞, 以避免意外排除工程化的EVs或在各種細胞培養條件下生產的EVs。
總之, ISEV建議使用通用術語”EV”和該術語的操作擴展說法, 以取代去定義那些不一致且有時會產生誤導的術語, 例如難以確定生物發生途徑相關的"外泌體"(exosomes 和 ectosomes) 字樣。
關於可添加為”EV”首標的”運作術語”(表2), 如果根據大小、密度、分子組成或細胞來源等特徵分離出一種或多種EV的子分類, 則會繼續鼓勵而謹慎的使用。
例如”小”和”大”等術語通常用於表示過去幾年中的EVs群體, 指的是使用過濾或差速超速離心(差速UC, dUC)等方法分離出特定大小的EVs群體; 然而, 儘管”小”通常可能指直徑小於200nm的EVs, 但對於大小的上限或下限卻沒有嚴格的共識, 而且很明顯, 許多分離方法(如dUC)會產生大小分佈重疊的EVs群體。
因此, 雖然這樣的術語可能仍在使用, 但研究人員應該意識到它的局限性, 並努力盡可能清楚地來定義其術語。
如上所述, 與所推測生物發生途徑相關的術語應謹慎使用, 並應附上強而有力的證據; 術語中的exosome(外泌體)是指來自細胞內部隔室並通過多泡體(MVB)所釋放的EVs, 而術語“ectosome”(也稱微泡、微粒)指的是來自於細胞表面的EVs, 這邊使用了許多專門術語來表示在特定細胞過程中出現的EVs, 例如細胞遷移(migrasomes)或程式化的細胞死亡(apoptotic bodies)。
在某些情況下, 特定EV子分類中的生物發生或釋放可能會受到藥理學或遺傳干預的抑制或刺激(另見7.1); 不幸的是, 大多數的EV分離技術並不能富集到通過不同機制所產生的EVs, 基於生物發生的子分類之表徵確定也很困難, 因為沒有exosome、ectosome或其他EV亞型的單一分子標記。
因此, ISEV不鼓勵使用基於生物發生的術語, 除非對這類EV群體進行了特別的分離和表徵方法。
值得注意的是, ”sEV”(用於小EV)和”exosome”(外泌體)並不是同義詞, 因為小EV群體包括了exosome和ectosome; 由於上述原因, 大多數現有的”exosome”和“ectosome/microvesicle”文獻都涉及到了廣泛的EVs群體, 而非通過特定生物發生途徑釋放的EVs。
某些類EV粒子可能不完全符合上述EVs的定義, 例如, 如果一個細胞被擠壓後所產生的粒子就沒有嚴格地遵守從細胞中”釋放”的定義。
2.2 EV模擬物 (EV Mimetics)
“EV模擬物"(EV Mimetics, EVM)等術語可用於表示通過細胞直接破碎、由分子成分從頭合成或通過天然EVs與例如脂質體融合所產生的EV類粒子。
無論此類粒子使用何種命名法, 它都將代表的是一般的生產過程, 其粒子與天然EVs的粒子有所區分, 並且不會聲稱其與來自特定生物發生途徑的EVs相似; 也就是說, 避免使用”exosome-like vesicles”和錯誤暗示特定生物發生的相關特性之類似術語。
2.3 非細胞外囊泡粒子 (NVEPs)
越來越多的人意識到, 多樣性的非細胞外囊泡粒子(Non-vesicular Extracellular Particles, NVEPs)通常會與EVs一起共同被分離出, ISEV學會特別被要求在MISEV2023的階段提供關於如何處理和命名這些粒子的導引。
由於ISEV是EV的專家學會, 因此不能擅自為其他類型的細胞外粒子建立命名法, 例如脂蛋白粒子(LPPs)、核糖核蛋白粒子(RNPs)、病毒或各種新提出的粒子類型, 如: exomeres和supermeres(外泌超小體)。
然而, EVs如何與其他粒子相關聯, 以及如何將它們與其他粒子分離並在複雜的混合物中與其他粒子一起表徵, 這些都與EVs領域密切相關。
因此, MISEV2023提供了以下的術語建議, 同時認識到可能需要其他的術語來提高清晰度(Figure 1, table 2)。
Table 2. EV命名和相關述語的快速參閱卡
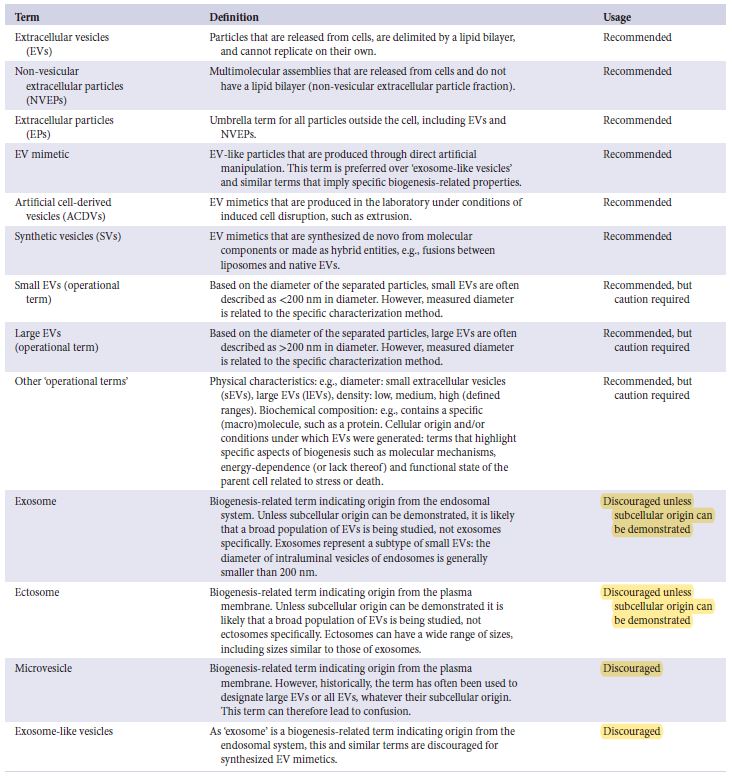
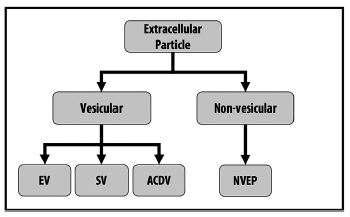
Figure 1. EP命名的階級。細胞外粒子包含了囊泡和非囊泡粒子。此表說明了在EPs類別之間的幾種區別和可能的命名方式: 細胞外粒子(Extracellular Particle, EP); 細胞外囊泡(Extracellular Vesicle, EV); 合成囊泡(Synthetic Vesicle, SV); 人工細胞衍生物囊泡(Artificial Cell-Derived Vesicle, ACDV); 非囊泡細胞外粒子(Non-Vesicular Extracellular Particle, NVEP)。也可見Section 2和Table 2。
細胞外粒子(Extracellular Particles, EPs)是細胞所衍生由奈米至微米大小的多分子組裝體首選術語, 包括EVs和非囊泡狀實體。
非細胞外囊泡粒子(Non-vesicular Extracellular Particles, NVEPs)是由一種或多種分子類別(如蛋白質、核酸)之細胞衍生組分所形成的所有非EV粒子; 例如脂質(如果存在的話)就不會形成一個有所界定的雙層膜; NVEPs和EVs可能會具有重疊的物理化學性質, 並且在生物基質中的NVEPs數量可能會大大超過EVs。
因此, 大多數EPs的分離方法都會導致NVEPs和EVs共分離存在, 同樣的, 許多EPs的表徵方法並不能特異性的識別到EVs; 小於EVs的NVEPs可能會無法經由某些EV表徵方法而檢測到, 因此其在EPs製劑中的數量可能還仍然未知。
因此, 當EVs和NVEPs還不能完全區分時, 術語”EPs”可能是適用的, 或者使用"EV製劑"或"含EV的製劑"。
建議:
1. “細胞外囊泡”是指由脂質雙分子層包裹且不能自行複製的粒子。
2. 鼓勵使用表定的運作術語, 但也要注意因為這些術語可能會受到其分離方法的影響。
3. 除非能夠證明特定條件和EV的亞細胞來源, 否則不建議使用生物發生的術語。除了少數的例外情況之外, 代表所研究的是廣泛之EVs群體, 而非特定的ectosomes或exosomes。
4. “細胞外粒子”是細胞衍生奈米至微米尺寸範圍的多分子組裝體總稱, 包括囊泡狀和非囊泡狀實體。
5. “非細胞外囊泡粒子”是一個準確的術語, 指本質上是非囊泡狀的細胞衍生多分子組裝體(即細胞外顆粒的非囊泡狀部分)。
共識: 79.5%(793)的MISEV2023調查受訪者”完全同意”, 19.9% (199)同意第2節:術語中的”大部分”。0.4% (4)”大多數”不同意, 0.2%(2)表示沒有意見和(或)專業知識; 沒有受訪者“完全”不同意。
以上內容即非經許可請勿任意複製轉發分享, 如有任何建議或詢問, 歡迎和我們聯繫
4. EV 分離與濃縮法
細胞外囊泡(EVs)通常在經過一次或多次分離或濃縮程序後會進行表徵和測試。國際細胞外囊泡學會(ISEV)先前已評估了這些方法的趨勢(Gardiner et al., 2016; Royo et al., 2020)。分離和濃縮方式可以根據 EV 的生物物理特性, 如大小、密度、電荷和表面組成(特定表面分子)來進行。
有時用於這些程序的其他術語包括「富集」(enrichment)、「純化」(purification)和「提取」(isolation)。
分離/濃縮後獲得的材料是「含 EV 的製劑」(EV-containing preparation)或「EV 製劑」(EV preparation), 這可能會需要在分析或使用前進行儲存的動作。任何分離方法都應根據特定 EV 來源的已知特性以及所需的 EV 產量和特異性來選擇。
在分離複雜的生物流體時, 總 EV 的產量和特異性之量化很可能只是估計值, 因為顆粒數量的量化並不全是具有 EV 特異性的, 並且/或通常依賴於 EV 豐度的替代指標, 例如尖峰群體(spike-in populations)或可檢測亞群的測量。
圖 2 顯示了一些常用的 EV 製備方法在產量(回收率)和其特異性對比的位置。本節提供了關於其中一些方法的信息和建議。更詳細的信息可以在文獻中找到(Hendrix et al., 2023)。
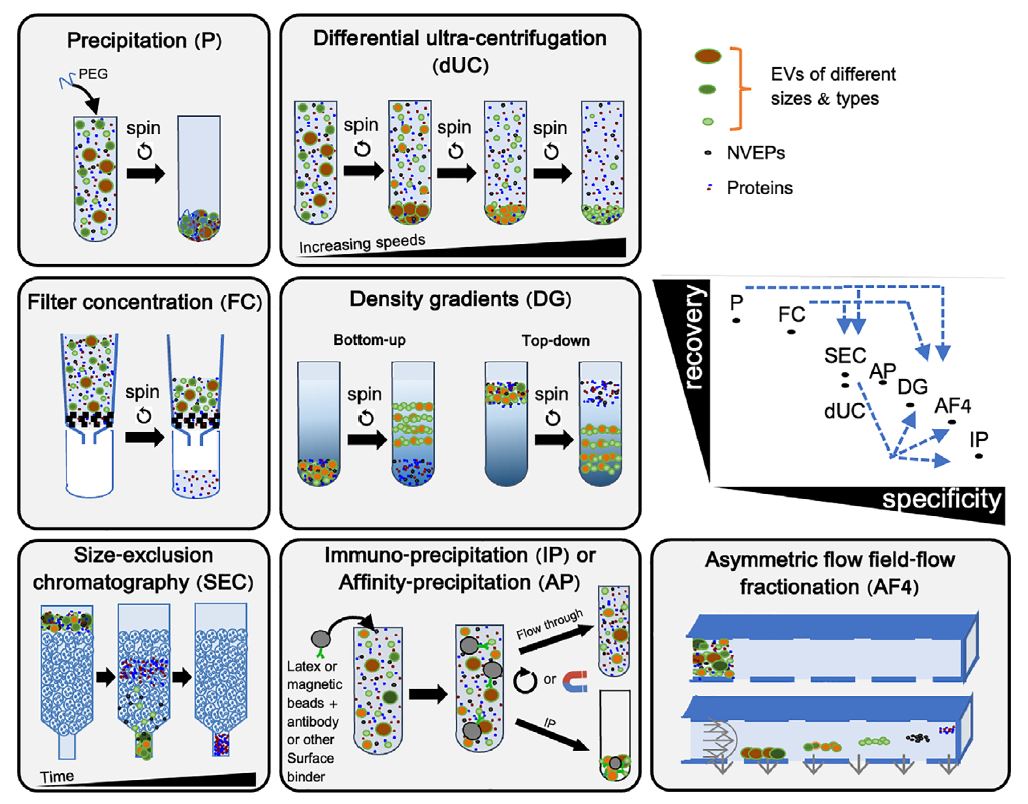
圖2: 主流的細胞外囊泡 (EV) 分離和濃縮方法在回收率(產率)和特異性關係圖上的位置。可提高特異性的方法組合; 特異性可以分為不同類型: 尺寸排阻色譜法 (SEC) 會根據粒徑將 EV 和許多(但並非全部)的非囊泡外泌體 (NVEP) 分離, 但所有類型的 EV 都會被一起回收;而差速離心 (dUC) 則會根據粒徑/重量分離 EV 的亞型, 但也會高速分離出 NVEP。請注意, 許多「外泌體純化」的試劑盒使用的是沉澱法 (P), 因此無法分離出乾淨的外泌體, 甚至無法分離 EV, 而是分離出包含有多種外泌體的混合物;而有些試劑盒則使用親和沈澱法 (AP), 該方法可能對 EV 的特異性更高, 但對外泌體的特異性較低。開發新方法的研究人員應考慮將他們的 EV 分離結果定位到這樣的圖表中。
4.1 EV的濃縮法
EV 研究中的濃縮是指增加顆粒的數量與樣本體積的比值, 在某些情況下可能需要進行濃縮。對於 CCM(細胞上清液)、尿液或牛奶等大體積的原始樣本, 可能需要在 EV 與其他 EP進行分離純化前進行濃縮的步驟。
例如: 色譜管柱可能有最大的上樣體積, 而如果原樣能首先進行濃縮的步驟(例如某些免疫分離程序), 搭配這樣的分離方法可能會更有效, 濃縮的方法可能會但不一定會實現某些程度的顆粒類型分離。
濃縮可以通過幾種方法完成: 如果是採用基於聚合物的沉澱方法則會減少了生物分子對溶劑的可用性而將水分子「擠出」, 這使得懸浮/溶解的材料(包括 EP)可以通過低速離心沉澱。
一些被描述為「外泌體分離」試劑盒的商業試劑盒實際上是採用了這種聚合物的沉澱法, 並沒有嚴格的「分離」出乾淨的EV顆粒, 更不用說 EV 的亞型了, 沉澱方法可能無法實現 EP 的任何明顯分離。
在過濾的方式中, 懸浮液會通過過濾器例如以重力、離心或真空的方式: 液體和分子量只有小於過濾器截留值的分子會通過, 而大於截留值的 EP 則會在過濾器的濃縮液體隔室中被回收。
目前有多種過濾器的截留值可供選擇,包括 3、10、100 和 1000 kDa, 這使得過濾法能夠實現一定程度的粒徑分離而不僅僅是濃縮而已; 100 kDa 的截留值可以保留 EV顆粒同時去除許多的背景雜蛋白質, 而 1000 kDa 的截留值可能會讓一些較小的 EV 顆粒通過。
然而, 另一個考慮因素則是回收率, 因為不同的過濾器/管可能會允許不同程度的 EV「粘附」於過濾膜上從而影響回收率; 請注意, 過濾法也可能用於在前過濾隔室中保留微生物(滅菌)或大型的 EV/EP顆粒, 但應注意避免出現擠壓情形。
切向流過濾(TFF,也稱為錯流過濾)是一種以過濾器來進行的濃縮方法, 其液體和比孔徑小的分子會以垂直於施加到含 EV 液體的流動方向來通過過濾器。這可以讓連續流動和液體能進行重複的通過濾膜除非過濾器已發生堵塞, 因而能允許處理大體積的液體。
與其他的過濾方法一樣, TFF可以根據過濾器的分子量截留值來進行基於粒徑的分離, TFF 已成功且可重複地用於大規模 EV 生產, 例如用於治療的應用上。最後, 濃縮也可以通過(超速)離心法進行, 其參數將在下一節中描述。
總結: 濃縮的方式可以通過基於聚合物的沉澱、過濾(包括切向流過濾)和(超速)離心來完成。 根據具體方法和其變量”例如過濾器截留值(粒徑或分子量)”的不同, 其會產生含有不同量的 NVEP 和蛋白質的 EV 製劑。
報告的建議: 對於濃縮的流程報告內容如下: 用於濃縮的材料性質; 生物流體的初始和最終體積; 處理時間(聚合物孵育、通過過濾器或直接離心); 流速(對於 TFF); 粒徑或分子量截留值(對於過濾/濃縮); 濃縮過程中的溫度控制。
4.2 差速(超)離分離法
差速超離(dUC)的原理是對含 EV 的液體施加不斷增加的相對離心力(RCF = g 力), 其完整的供體細胞或組織已首先通過一次或多次低速離心以去除, 目的是按順序以沉澱沉降係數遞減的 EP顆粒。
由於球體的沉降速度與其直徑的平方以及顆粒與介質之間的密度對比成正比(斯托克斯定律方程), 最大和/或最密的 EP顆粒會傾向於在第一步(中速/短時間)中沉澱下來, 而最小和/或最不密的 EP顆粒則主要會在更高速度/更長時間的離心後被回收。然而在實踐中發現, 這種方法並無法實現完美的 EV 分離, 並且來自不同離心速度的沉澱物會具有重疊的特性以及可變的生化和物理參數。
無論使用何種離心步驟應如 MISEV2018 中所述, 需報告轉速(rpm)和轉子的類型(以允許計算調整後的 k 因子)、離心時間(以允許計算沉澱顆粒的沉降係數)和操作溫度, 還應報告設備的加速和減速設置。在文獻中所報導的典型 dUC 工作流程是施加約 10,000 至 20,000 × g 的最大力持續 10 至 90 分鐘以富集推測較大/較密的 EV顆粒, 而施加約 100,000 至 200,000 × g 的最大力持續 45 至 150 分鐘是用以沉澱推測較小/較輕的 EV顆粒。這些數字可用於計算通過這些不同方案回收的顆粒沉降係數(S): S = 轉子的調整 K 因子/離心時間。理論上S 係數在 15–150 範圍內的顆粒通過「較大 EV」的離心條件而被回收, 而 S 係數在 2 至 5 範圍內的顆粒通過「較小 EV」條件而被回收。可以通過延長離心速度和時間來回收具有較小 S 的顆粒, 代價是會增加 NVEP/游離蛋白的共分離現象。根據離心參數不同, 所得沉澱物可能會富集較大/較密或較小/較輕的 EV顆粒但無法將這些群體的進行完全分離。EV 的產量也可能較低尤其是在富含雜蛋白背景的液體(例如血液原樣和複雜的培養基成分)懸浮液時, 並且這個問題可能無法通過簡單的增加離心時間或速度來解決。
大多數已發表的研究都集中在較小的 EV 上而丟棄和/或不分析通過較低速離心獲得的沉澱物。然而為了讓研究之間可比較並避免沉澱較大的顆粒並可能會引入假訊號, 會建議執行某些初步離心。高速 UC 的強大 g 力也被證明其會誘導 EV顆粒的團聚(Linares et al., 2015), 但這可能不會在所有的 EV 來源中被發現。在分析新的 EV 原樣時應保留中間離心的沉澱物並至少與最終的最高速沉澱物並排分析一次以確定所感興趣的分子或活性是否會特異性地富集在小 EV 顆粒中或者是存在於其他亞型中。
總結: dUC法會富集根據其沉降係數所分離的 EV 亞型, 沉降係數與其粒徑和密度成正比並會共分離出與 EV 具有相同沉降係數的 NVEP顆粒, 尤其是在高速和長時間超離心後會可能誘導 EV顆粒的團聚。
報告的建議: 對於差速超離法應報告這些內容: 速度、轉子類型和離心時間, 以允許計算調整後的 k 因子(適用於其他轉子)和沉澱 EP 的沉降係數; 轉子類型和離心管內的樣本體積; 離心過程中的溫度; 加速和減速(制動)的設置。
4.3 密度梯度分離法/墊層分離法
密度梯度法或墊層可用於根據不同類別 EP 的特徵密度而將某些 NVEP 和背景雜蛋白與 EV顆粒進行分離。梯度由不同比例的選定緻密介質(如蔗糖、碘克沙醇或碘海醇)和水性緩衝液所組成的墊層所製備, 密度從梯度底部到頂部而遞減, 而墊層由緻密材料的均勻層組成, 位於水性柱的下方。
含 EV 的原樣可以裝載在梯度下方(「自下而上」)或裝載到梯度或墊層的頂部(「自上而下」)然後進行超速離心。在自下而上的方法中, 含 EV 的原樣會與高密度的介質混合後裝載在離心管底部並覆蓋有密度遞減的墊層;原樣也可以鋪設在製備好的梯度下方。隨著超速離心進行, 密度小於周圍介質的顆粒會向上漂浮。在足夠的時間內, 顆粒最終將到達與其浮力密度相對應的密度分數中。
由於較小的 EV 移動速度會比大型 EV 慢, 尤其是在具有粘性的介質中, 因此對於蔗糖密度梯度UC法中的自下而上方法也可用於根據粒徑來分離 EV。
在自上而下的設置中, 低密度介質中含 EV 的原樣會裝載於梯度或墊層的頂部:
對於梯度來說, 顆粒以與其密度和粒徑相對應的速度來進入梯度, 直到達到其平衡浮力的密度。
對於墊層來說, 到達墊層的顆粒如果密度小於墊層材料則會保留在其界面處。
但如果密度更大的話則會繼續進入並穿過墊層。因此, 墊層的方法會更容易實施來通過密度閾值以分離出EP顆粒。
重要的是對於梯度來說可能會需要長時間的超速離心才能實現最佳的分離效果[例如超過 48 小時], 但較短的離心就可能足以滿足某些應用[例如,1-2 小時, 16 小時]。通過梯度分離後, 必須仔細收集餾份以避免破壞梯度。
確認最終級分的密度是非常好的做法, 例如通過稱重給定的體積或測量其折射率, 在執行大多數下游測定之前必須去除密度介質, 這可以通過例如用緩衝液稀釋餾份並進行超速離心或接續使用SEC法來完成, 密度梯度和餾份洗滌後的回收率會相對較低。
總結: 密度梯度和墊層可以根據目的而在不同的設置中實施(自上而下、自下而上),將 EV顆粒與背景雜蛋白分離或與 NVEP進行分離或分離出 EV 亞型。這導致會得到高純度的材料(基於密度)而回收率低。
報告的建議: 對於密度梯度和墊層應報告這些內容-- 密度材料、緩衝液組成和梯度/墊層製備的確切方法; 裝載材料的體積和濃度以及裝載到柱頂部或底部的方法; 離心參數的詳盡描述(與 dUC 法相同); 收集程序、餾份的最終密度(如果相關)和洗滌的細節。
4.4 尺寸排阻色譜分離法
尺寸排阻色譜法(SEC)是根據粒徑來分離包括 EV 在內的奈米顆粒(Boing et al., 2014; Karimi et al., 2018)。在 SEC 中, 樣本會被放置在裝載包含具有特定孔徑通道的基質的柱頂部, 在重力或泵的壓力驅動下, 較大的顆粒會快速的通過基質而不會進入孔隙中, 這可以做為較早期的餾份收集; 而較小的顆粒(小於基質孔徑)其留滯的時間更長並主要會在較晚的餾份中洗脫出來。
某些SEC基質可允許將符合EV 粒徑的顆粒(EV、病毒、較大的脂蛋白顆粒)與較小的 NVEP 和游離雜蛋白分離出來, 影響SEC分離程度的變量包括基質的組成、孔徑和管柱的填充方法、管柱的長度與直徑(或體積)的比值、流速(重力與給定的壓力)以及施加的樣本濃度和體積。
尺寸排阻色譜管柱可以是自製的或購買的, 商業的管柱通常會在嚴格控制的條件下填充並且可能會比自製管柱具有更高重複性的分離結果, 與所有其他方法一樣, 這必須通過仔細的表徵來確定收集餾份中的 EV 和其他 NVEP 之豐度和純度。
SEC分離法會稀釋樣本而與輸入的原樣相比增加了體積, 因此可能需要在SEC分離法之前或之後對樣本進行濃縮體積, SEC分離可以通過修改基質與結合親和的方法來優化。
相關的鍵結洗脫色譜(bind-elute chromatography)方法會將基於粒徑的分離與基於電荷或分子親和力的選擇方法相結合並允許單次的流洗方式(保留不需要的部分), 這可能會適用於高通量分離應用中例如運用在多孔盤配件。在某些情況下SEC基質可以在徹底清潔後重複使用。
總結: SEC是一種易於操作的基於粒徑的顆粒分離技術。管柱可以填充各種基質並以不同的規模進行, 具體取決於所需的分離容量和分辨率。
報告的建議: 對於SEC報告應包含這些內容-- 基質類型和孔徑; 含基質柱的高度和直徑(或體積); 管柱填充方法(或商業上可獲得管柱的來源); 通過SEC前的樣本來源、通過體積和顆粒濃度, 包括任何預先的分離/濃縮步驟; 緩衝液組成; 指定重力流或壓力。如果是壓力需指明泵的系統和壓力參數; 空隙體積以及收集的餾份數量和體積; 通過SEC後的任何濃縮方法; 如果管柱被重複使用則需要包含管柱的清洗方法和管柱的使用次數。
4.5 基於流體的流動分離法
基於流體的流動技術是根據一個或多個顆粒特性來分離 EV 與其他顆粒, 其不依賴於「基質」或固定相。目前用於 EV 研究的兩大類流動技術是場分餾(FFF,Giddings et al., 1976)和自由場電泳(FFE,Preußer et al., 2022)。這些技術可以應用於高度異質的輸入原樣並且沒有固相存在所以可有高的顆粒回收率, 這也是最溫和的分離方法之一因此可用於研究或與 EV 顆粒相關的分子中。
EV 研究中最流行的 FFF 方法是不對稱流 FFF(AF4,Sitar et al., 2015), 樣本中的顆粒會通過一個長而薄的通道以拋物線模式的流體流動進行傳輸, 而垂直於傳輸方向的場會傾向於將顆粒濃縮到通道底部。
擴散更快的較小顆粒更有可能會進入通道中間更高速度的流動區域, 因此顆粒可通過流體動力學來實現粒徑分離。一定程度的純化也可以通過由分子量截留的過濾器組成的通道底部來實現。
儘管 AF4 可以精確地分離粒徑差異很小的顆粒群體但在其標準配置中它不是 EV 特異性的; AF4 也不是一種製備技術。相比之下, FFE方法是將流動與電泳相結合而增加了例如等電點的分離, 在分離通道中引入具有不同 pH 或其他特性的分離緩衝液來允許其對不同的 EV 和其他 EP 群體進行高分辨率的分離。FFE 可以以各種規模進行。
總結: 基於流體的流動分離例如 AF4可以實現高分辨率的基於粒徑的分離。缺乏固相且施加的力有限, 流動會比大多數 EV 分離技術更溫和。基於粒徑的分離可以通過施加不同類型的場與基於其他原理的分離相結合。製備規模可以通過自由場電泳等流動技術達到。
報告的建議: 對於基於流體的流動分離需報告這些內容-- 儀器的所有訊息包括泵和其收集裝置; 所有緩衝液的組成及其過濾流程。特別是在 FFE 中, 如何確認緩衝液的特性; 所有場的特性例如流速和壓力、pH 梯度、電場; 分離室的粒徑和組成包括任何分子量截留板; 餾份收集的所有相關細節。
4.6 基於電荷和分子識別的分離法
所有的親和方法共同原理是根據 EV 的表面電荷或分子組成來捕獲 EV。離子交換色譜是利用帶相反電荷的顆粒/表面之間非常簡單的親和力: EV 和/或 NVEP會根據負(陰離子交換)或正(陽離子交換)表面電荷對基質具有親和力的作用來分離。
相比之下, 分子生物學中常用的「親和分離」一詞指的是利用一種大分子複合物對另一種大分子複合物的特異性識別的方法。在這種情況下, 親和探針包括肝素和各種凝集素會對特定脂質或蛋白質且具有親和力的特定全長蛋白質讓其結合特定 EV 表面的蛋白質或更普遍地結合膜上的肽, 包括選擇特定粒徑範圍內 EV 的曲率感應肽; 稱為”適配體” [為結合特定靶標而開發的短單鏈 DNA 或 RNA 分子]。為識別特定 EV 表面分子而產生的抗體是最常用的親和試劑, 其最常用的靶標是四跨膜蛋白(tetraspanins)。
在基於分子識別的親和方法中, 含 EV的原樣(可能已根據第 4.1 節進行濃縮)在親和探針結合到基質(例如膜或珠子)之前或之後的過程中就會被引入。若是珠子則反過來可以放置在柱子或管子中以促進結合和洗滌。
分子靶標展示在原樣會通過親和探針結合到基質上(「下拉」), 而未結合的部分則會流走。非特異性結合的材料可以通過一次或多次洗滌去除。如果 EV 是預期的標靶物則於稀釋和洗滌緩衝液中不應該存在去污劑, 除非濃度非常小(0.001% 或更少), 以最大限度地減少對捕獲基質或 EV 之間的非特異性結合出現。為了評估靶向 EV 回收的效率和特異性, 建議至少在方案開發期間, 通過生化分析並排比較流穿和下拉情形以測量親和基序和一些 EV 標記(見第 5 節)。
結合的 EV 可以通過各種技術從基質中解離和回收, 範圍從改變緩衝液的特性到添加過量的靶分子(例如糖或脂質)再到消除有效結合所需的因素(例如使用 EDTA 螯合鈣)。
然而現實中有一些親和試劑可能會與其 EV 標靶緊密結合, 這需要通過例如蛋白酶來去除。在某些情況下, EV 結合分子和/或基質可能會與 EV 一起被回收而造成純度不佳, 如果下游分析不受這些材料的影響(例如被基於蛋白質的親和探針污染的 EV 的核酸分析), 這可能就不是個問題, 但在其他情況下可能會是有疑慮的(例如在功能測試中, 因為 EV 表面被 EV 結合分子所被修飾), 其抗體會特別難以和 EV 分離。傳統上用於分離抗體和抗原的低 pH 處理可能會影響 EV 的結構, 用蛋白酶處理也可能會消化掉 EV 表面上的蛋白質而造成結果失準。
在任何分子親和方法中, 重要的是要了解靶分子與 EV 相對於 NVEP 的關聯程度, 或與一種 EV 亞型相對於其他亞型的關聯程度並評估捕獲試劑的特異性。例如使用 CD9 或 CD63 親和捕獲尿液中的 EV(uEV) 就會排除了來自近端腎小管細胞的 uEV。另一個例子是關於 L1CAM 親和力(一種推定的神經元 EV 標記)的文獻自 MISEV2018 以來就已有了實質性發展, 儘管 L1CAM 已被研究作為一種膜相關抗原,用於從外周血原樣中分離出推定的神經元 EV顆粒, 但最近它被描述為在某些 EV 來源中以裂解的、大部分可溶式的形式存在。它也存在於來自各種來源的 EV 上而不僅僅是神經元所特定, 並且廣泛使用的抗 L1CAM 抗體也可能會識別到其他的標靶。
總結: 基於電荷和分子識別的親和方法是根據表面電荷或特定分子決定簇的接觸分離 EV 製劑的組分。將共分離所有接觸到特定電荷或分子決定簇的 EV 亞型或 NVEP, 特異性和回收率則取決於所選分子決定簇接觸的特異性與普遍性。基於抗體的親和分離會導致決定簇接觸的EP與抗體和/或分離珠子的共分離現象。在建立方案時必須通過量化下拉與流穿中回收的材料來評估其效率和選擇性。
報告的建議: 對於基於親和力的分離法應報告這些內容-- 用作親和探針的分子(性質、來源); 基質(珠子、凝膠、管柱); 孵育時間; 緩衝液和洗滌次數; 洗脫過程(例如洗脫緩衝液組成、時間)。
4.6 一般考慮事項和基於試劑盒方法的注意事項
由於 EP 會有重疊的生物物理特性以及各種 EV 來源中許多 NVEP 的豐度差異, 越來越多互補的分離技術會依序採用(圖 2 中的箭頭處), 這可以增加其特異性, 用於將 EV 與蛋白質聚集體和其他 NVEP 分離的方法示例包括尺寸排阻色譜、密度梯度超離、不對稱場域分餾(AF4)和超高速超離心。
然而其中一些研究表明, 先前被提議為 sEV 標記的幾種蛋白質在 NVEP 中同樣(如果不是更多)會表達豐富, 因此需要重新評估所出現的 EV 選擇性。
與此相反的是, 自 MISEV2018 以來一個日益增長的認識是, 一些與 EV 會共分離的分子包括蛋白質、核酸、糖和脂質, 可能不被視為「污染物」而是被當做是動態 EV「冠」(EV ‘corona’)的一部分。分子甚至生物奈米顆粒例如脂蛋白可能會吸附到 EV 表面, 在那裡它們可以作為生物標誌物或有助於 EV 的功能。
EV 冠可以通過某些分離過程(包括 dUC 和 SEC)達到部分或完全去除, 正在進行的 EV 冠研究可能會改變我們對污染物和對高度純淨 EV 顆粒感知需求的看法; 這一點也與 MISEV 的下一節(EV的表徵)相關。
本節僅描述使用容易獲得(即商業上)的設備和儀器的方法。然而, 分離方法的新發展包括涉及在單個實驗室中構建設備的方法正在不斷發生並受到 ISEV 的大力鼓勵。在建立新的分離/濃縮工作流程時, 一個好的做法是使用第 5 節和第 6 節中討論的方法以及仔細和完整的記錄來評估 EV 分離/濃縮的程度。還建議將結果與另一種已建立的方法的結果進行比較。例如可以跟踪 EV 標記蛋白並將其與總分離蛋白相關聯, 以確定相對於報告的總蛋白的富集倍數並解釋 EV 標記損失, 其結果將表明回收率和富集程度並且還將顯示分離的 EV 群體是否仍代表其原始群體或已被選擇性的獲取。
最後是關於商業試劑盒的一些注意事項, 許多試劑盒被宣傳為可獲得特定類型的 EV(通常是「外泌體」)或來自特定類型來源的 EV。這些試劑盒可能會或可能不會基於各種原理(包括聚合物沉澱、膜親和力、抗體捕獲和過濾)來實現 EV 的分離或濃縮。這些試劑盒在某些情況下可能有用但 EV 研究人員應注意幾個主要的警告: 如不披露 EV 分離/濃縮原理細節的試劑盒可能會產生難以解釋的結果, 尤其是因為它們可能會引入未知的污染物(例如某些聚合物沉澱試劑盒的聚乙二醇), 這可能需要額外的工作將這些方法與其他技術的結果進行比較並將結果定位在回收率/特異性圖示上(圖 2)以獲得更好的解釋。特別是基於沉澱的試劑盒會濃縮混合物中的所有 EP與非常多的游離蛋白質並導致高度不純的製劑; 尤其是來自複雜的且富含 NVEP 的來源樣本例如血漿和血清, 強烈不鼓勵使用此類試劑盒除非僅用於減少體積。
相比之下, 基於親和力的方法可能會只分離出 EV 的亞型, 如果未披露確切的試劑類型則會難以評估親和試劑的特異性。通常應優先選擇已完全披露內容和原理的試劑盒而不是那些提出未經證實的聲明(例如「外泌體」分離)且不提供細節的試劑盒。
以上內容即非經許可請勿任意複製轉發分享, 如有任何建議或詢問, 歡迎和我們聯繫
5. 關於EV 的表徵
EV 的表徵對於估計 EV 數量、確定 EV 的存在以及評估非 EV 組分對 EV 製劑的貢獻是必需的。表徵面臨的挑戰包括:粒子粒徑小、EV 粒徑和分子的異質性, 目前缺乏單一種通用的 EV 識別方法和許多測量技術所包含的非 EV 特異性。
因此目前並沒有單一的測量或方法能夠滿足所有 EV 表徵要求, 在此建議要使用正交的方法(即沒有相同測量限制的方法)。
如果需要對 EV 製劑提出要求, 那為了證明這些要求就需要對樣本要進行表徵的程度可能會取決於材料的來源(參見第4章節)。
這可能意味著需要對不同的樣本進行額外的表徵步驟, 也可能意味著需要額外的報告訊息以便評估其他分析前的變數對 EV 的影響。
EV 的總體組成(對蛋白質、脂質、核酸和其他生物分子總質量的貢獻)會因 EV 的來源而不同。雖然測量這些單顆分子分類可用於估計 EV 的豐度但這些數值並不一定會與 EV 的濃度是完全相關並且在不同來源的材料之間也沒有其普遍性;因此它們不應用作 EV 濃度的單一測量標準。
正如沒有單一的分子類別測量可以量化所有的 EV 一樣, 同樣也沒有 EV 或 EV 亞型的通用分子標記物。
標記物必須根據其來源和類型的特異性證據來做選擇。無論其來源為何, 目前為止尚無已知的通用標記物可以識別所有 EV。
儘管已提出了數種蛋白質作為 EV 生物發生途徑的推定標記物(例如Annexin A1、SLC3A2 和 BSG)用於所推測的外泌體(ectosomes)中以及 Lamp1用於推定的外泌體(exosomes)中, 然而這些標記物的普遍性尚不清楚或未被接受。
請注意, 涉及四跨膜蛋白 CD9、CD63 和 CD81 之基於親和力的方案並非對做為 EV 亞型的外泌體具有其特異性; 使用針對這些四跨膜蛋白中每一種抗體富集的 EV 群體在分子組成上並不是完全重疊的。
此外, 並非所有的 EV 都顯示出這些四跨膜蛋白, 因此四跨膜蛋白富集並不能捕獲所有的 EV 顆粒。
對相同參數進行正交的方法測量不太可能會具有相同的偏差值; 例如從光學方法與非光學方法來推導粒徑大小。
使用正交的方法來對 EV 樣本進行表徵其對提供證據於證明共同分離物不是生物標記或功能發現的原因是至關重要的。
由於許多 EV 的表徵方法要麼不具備 EV 特異性或要麼無法檢測出所有的 EV, 因此需要透明的報告其分析方法和結果以實現 EV 數據的可重複性。
先前已開發並更新了 EV 數據報告框架即為 EV-TRACK, EV 表徵的標準化得到了 ISEV 研討會、ISEV 嚴謹性和標準化工作組以及 ISEV 立場文件的支持。
在以下各節中將討論 EV 表徵的不同方法, 每節都提供了採用該表徵方法時的建議。無論採用何種方法, 表徵的總體建議總結如下:
建議事項
-
每個 EV 製劑都應通過 EV 來源的定量測量來定義(例如: 分泌細胞的數量、生物液體的體積和組織的質量)。
-
應對 EV 的豐度進行大致的估計值(顆粒數量、蛋白質和/或脂質含量)。
-
應測試 EV 製劑中是否存在與 EV 亞型或一般 EV 相關的組分, 具體取決於所希望達成的特異性。
-
確定非囊泡與共同分離組分所存在的程度。
-
當使用定量指標對 EV 進行表徵時需提供儀器/方法的偵測極限 (LOD) 指示。
5.1 粒子數量濃度的量化
EV的數量可以和體積的測量一起用於定義其數量濃度(以粒子數/毫升為單位), 這是一個被廣泛報告且用於檢測輸入標準化、檢測結果測量和體內給藥的指標。
然而因為許多技術是缺乏對 EV 的特異性和對所有 EV 的靈敏度導致其通常並不可靠。
ISEV 嚴謹性和標準化 EV 參考材料工作組最近概述了測量技術中的考慮因素以及該領域在邁向可追溯測量方向面臨的挑戰, 可用於開發和報告具有良好特徵的 EV 參考材料。
這項工作的一個關鍵亮點是需要報告檢測的LOD(偵測極限)值, 這可允許其他人在不考慮靈敏度限制的情況下驗證其結果。
請注意, 根據測量方法中所報告的血漿內 EV 的濃度會跨越六個數量級。
因為正交的方法不會共享相同的測量極限, 通過使用正交的方法是可以提高 EV 濃度測量的信心, 每種方法都有特定的 LOD(偵測極限)值, 例如檢測光散射的強度、螢光的強度和物理性粒徑。
例如對於使用粒徑標準品確校的電阻脈衝感應(RPS)技術就可以報告粒徑的LOD(測量極限)值。RPS技術的較低LOD(測量極限)值很可能是由於靈敏度的限制, 而較高 LOD(測量極限)值則將會受到孔徑的影響。
對於流式細胞術等光學技術, LOD(測量極限)值可以報告為直徑(源自光散射光學模型)或等效的可溶性螢光團分子(MESF)(源自螢光強度)。
這些方法可在儀器與靈敏度觀點產生一致的數據, 目前由於涉及粒子可檢測性的變數數量, 並沒有方法可以為奈米粒子追蹤分析 (NTA)、DLS 或影像式流式細胞術推導的可追溯LOD(偵測極限)值。
沒有使用任何表型表徵但可以輸出濃度測量的技術例如使用膜染料, 可能會由於染料自我聚集以及無法區分 EV 和其他共分離物而導致 EV的濃度數據被高估, 無論其成分和來源如何, 除非其可普遍染色到所有的EV否則缺乏這些問題的膜染料則可能會導致低估濃度數據, 而目前尚未報告過此類染料。 更進一步的儀器和檢測特異性建議可在第 6 節中找到。
對於無法區分 EV 與其他潛在共分離物或懸浮污染物(例如蛋白質聚集體)的技術, 無論採用什麼上游的分離步驟都建議將濃度報告為「粒子或 EP 濃度」而不是「EV 濃度」。
建議事項
-
報告每個檢測的 LOD(偵測極限)值或說明其不可量化或未知。
-
在可能的情況下需報告稀釋系列的數據以證明濃度推導位於系統測量的線性區域中。
-
在可能的情況下應使用正交方法確定粒子數量。
-
除非方法對 EV 具有高度特異性否則這些測量的結果應描述為與「粒子」或「EP」相關。
5.2 粒子粒徑大小的量化
EV 粒徑(以奈米半徑或直徑計)的測量依賴其假設於例如球形度或移動性並且結果可能會受到上游變數的影響。 常見的高通量方法是假設 EV 為球形的包括流式細胞術、NTA、RPS、多角度光散射和動態光散射(DLS)技術。
雖然「粒徑」和「直徑」在測量方法之間經常會互換採用, 但它們的推導方式也會導致測量技術之間存在有一致的差異。例如依賴於粒子移動性的技術, 比如 NTA 或 DLS其所測量的是流體動力學直徑, 這與諸如冷凍電子顯微鏡(cryo-EM)等成像方法相比會導致粒徑值的高估。
很少有方法能夠在整個可能的 EV 直徑範圍內(從數十奈米到微米)準確地測量 EV 粒徑。例如雖然 cryo-EM 的高解析度成像是最準確的方法之一但其通量相對較低, 並且許多豐度往往低幾個數量級的較大 EV 可能會無法被量化, 而量化低對比度且小於 100 nm 的 EV 能力也可能是一個限制因素。
隨著越來越多的研究人員使用具有更高靈敏度的專用單顆粒技術, EV 粒徑分佈是異質性的且通常是多模式的, 其分佈取決於 EV 的來源(例如細胞類型、組織、生物液體)和分離方法(例如超速離心、尺寸排阻色譜、沉澱)。
這種異質性突顯了報告完整粒徑分佈的重要性而不僅僅是單一指標(例如平均或中位數直徑)。
建議事項
-
報告 EV 製劑的完整粒徑分佈(例如直方圖或密度圖)。
-
報告粒徑分佈的平均值、中位數和標準差。
-
在可能的情況下需使用正交方法確定粒子粒徑。
-
如果是報告單顆粒徑指標, 請說明選擇該指標的原因。
5.3 總蛋白質的量化
EV 製劑中的總蛋白質(以 μg 或 μg/mL 濃度計)可以通過比色法、螢光法、SDS-PAGE 上的整體蛋白質染色或吸光度讀數來估計,每種方法都有不同的靈敏度和準確性。作為一種批量分析技術,特別是對於特異性較低的 EV 分離方法或複雜的生物液體來源, 總蛋白質定量通常會由於共分離的蛋白質存在而高估了 EV 的濃度。相反的如是高度純化但產率低的 EV 製劑可能會對檢測的靈敏度形成挑戰。由於測得的蛋白質濃度可能會因測量到完整或被破壞的 EV 而異, 因此應指明物理破壞的細節以及任何去污劑的性質和濃度。
蛋白質濃度作為 EV 濃度的替代品應謹慎使用, 通常不建議使用因為每個 EV 的某些蛋白質富集可能發生在不同的細胞表型或刺激下。
由於蛋白質/粒子的比率也取決於每個檢測/儀器的LOD偵測極限值(較低的濃度檢測極限或較低的粒徑檢測極限), 因此如果是報告比率值, 建議要單獨提供絕對的蛋白質和粒子濃度。
建議事項
-
除非上游處理的證據對 EV 具有高度特異性,否則應按「粒子」或「EP」報告輸出。
-
報告每個檢測的較低濃度檢測極限以方便解釋。
-
對於比率值應報告原始組成的測量值,而不僅僅是比率值而已。
-
蛋白質濃度應在參考曲線的線性範圍內這也應在報告內。
-
報告是使用完整的製劑還是已被破壞的製劑。
5.4 總脂質的量化
EV 樣本的總脂質量化可以通過比色法、膜嵌入染料的螢光、全反射傅立葉變換紅外光譜(FTIR)或層析法來進行。然而嵌入染料的方法和 FTIR 對少量 EV 的靈敏度可能會不足並且某些方法需要高度專業化的設備。
這些技術是否能檢測到所有獨立於脂質組成的 EV 仍然是未知, 由於共分離的 NVEP(例如脂蛋白)導致總脂質測量可能會高估 EV。
建議事項
-
考慮您的檢測的 LOD(偵測極限)值。
-
考慮共分離的 NVEP 可能會如何影響您的測量。
5.5 總 RNA 的量化
RNA 是一種經常研究的 EV 相關分子(參見第 6.5 節), 因此 EV 製劑的基本表徵可能包括總 RNA 量化作為品質控制組分或用於分析和功能研究中的標準化。
總 EV 內RNA 的量化可以通過毛細管電泳和其他方法完成。然而由於許多 EV 來源中存在大量更豐富的 EV 外 RNA,因此很難推薦使用總 RNA 作為 EV 濃度或純度的替代品。一些 RNA 的量化方法不能區分 RNA 和 DNA, 所選的分離試劑盒也被證明會影響到下游結果。
早期的 ISEV RNA 立場文件建議使用靈敏的技術例如 Agilent Bioanalyzer pico 晶片或 Quant-iT RiboGreen RNA 檢測以用於EV內RNA的量化,而不是用像 NanoDrop 這樣靈敏度較低的方法。
然而有一些核酸染料例如 RiboGreen對 RNA 的特異性不如對 DNA 的特異性。此外小 RNA 需要專門的 Bioanalyzer 試劑盒。其他的靈敏方法包括 Qubit microRNA 檢測試劑盒,它對小 RNA 具有高靈敏度。
用不含 RNase 的 DNase 進行預處理可能對準確的 RNA 量化有用,因為許多技術對 DNA污染是敏感的, 然而DNase處理可能無法完全去除所有的DNA污染。
建議事項
-
考慮您的檢測區分 RNA 和 DNA 的能力以及您所選方法的檢測極限。
-
報告樣本的任何預處理酶,例如使用 DNase。
5.6 EV 形態的表徵
目前對於較小的 EV最好使用高解析度成像技術來評估 EV 的形態,例如掃描式電子顯微鏡(SEM)、穿透式電子顯微鏡(TEM)、冷凍電子顯微鏡(cryo-EM)以及掃描式探針顯微鏡(SPM)包括原子力顯微鏡(AFM)。
遠大於光衍射極限(≳200 nm 直徑)的 EV 可以通過傳統光學顯微鏡進行評估, 這些技術不一定可以互補或能夠獲得可比較的圖像質量。
例如乾燥條件可能會導致 EV 會形成人為的杯狀, 這在水合條件下是看不到的, 至少在粒子水平上如果成像技術能夠同樣好的可視化到共分離的 NVEP則它們可以允許來評估EV的純度。成像技術通常受到低通量和基於視野選擇的潛在偏差限制 。
建議事項
-
無論成像技術如何都需報告所有實驗細節包括儀器、軟體版本、採集和分析設置、樣本製備過程、如何選擇成像區域以及相關的對照和校準資訊。更多細節可在第 6.4.1 節和第 6.4.4 節中找到。
5.7 通過蛋白質組成表徵 EV
由於 EV 的異質性, MISEV2023如同 MISEV2018所述, 不能推薦特定 EV 亞型的分子標記物。MISEV2023 推薦 MISEV2018 中引入的五組分框架以用於報告有關 EV 蛋白質含量的主張(表 3)。
第 1 類和第 2 類是評估 EV 特徵的存在, 第 3 類是評估常見污染物的純度, 第 4 類和第 5 類是提供有關 EV 可能的細胞內起源 (4) 或共分離物 (5) 的額外資訊。理想情況下,應顯示 EV 製劑中的標記物相對於未分蹓來源材料的富集或耗盡, 為了避免對應到分析哪些 EV 蛋白質產生的誤解和限制,MISEV2023僅提供了一些提名示例(表 3)。
其他推定的標記蛋白可以使用諸如 Uniprot (https://www.uniprot.org/ ) 之類的資料庫以分配到其中一個類別, 其中「亞細胞定位」部分指示亞細胞區室或細胞外定位而「特徵」指示拓撲和跨膜結構域。 儘管這些類別可適用於 EV, 但無論分析的方法如何其中的一些標記物在某些單顆 EV 分析技術中可能無法在技術上使用,這可能還需要其他的對照比較。
還有多種方法可來確定蛋白質標記物的存在, 這些方法的靈敏度、特異性和可靠性可能有所不同, 當前的檢測和儀器特異性報告考慮因素在第 6 節中有概述。
建議事項
-
利用五組分框架(表 3)報告有關 EV 蛋白質含量的主張。
5.8 EV 的非蛋白質標記物
非蛋白質標記物例如磷脂酰絲氨酸、聚醣或特定核酸,這很少是 EV 特異性的, 但在某些情況下可能為脂質雙層或細胞質組分的存在提供額外支持。特別是對於單顆粒測量, 與蛋白質標記物的共定位也可能為 EV 的存在提供更有力的證據,例如膜嵌入染料和四跨膜蛋白陽性事件。
非蛋白質標記物可以直接通過脂質質譜或拉曼光譜等技術檢測(第 6.7 節),也可間接使用螢光探針例如膜標籤或腔內染料。 關於使用非蛋白質標記物標記 EV 的報告建議會在第 6.6 節中概述。
大多數非蛋白質組分標記物的非 EV 特異性促使我們應謹慎行事, 膜染料可能會與任何脂質複合, 包括 NVEP 的脂質;被腔內酶(例如酯酶)激活的染料可能不存在於所有 EV 製劑或亞型中;核酸染料已用於EV中,但仍需要關於對照和特異性的建議 。
建議事項
-
如果使用非蛋白質標記物,應同時考慮使用蛋白質共定位。
5.9 EV 相關組分的定位
EV 的相關組分例如蛋白質、核酸和聚醣, 可能是在腔內的、在膜中的或在EV 外部的, 拓撲學知識對於理解生物學可能會很重要。例如 EV 是否必須與受體細胞融合才能遞送腔內攜帶物, 又或者 EV 是否可以簡單地將表面相關分子呈遞給受體呢?因此應通過採用或改編先前發表的方法通過執行溫和消化、透化或親和試劑可及性來確定所推定的活性組分定位。
建議事項
-
考慮如何在方法設計中確定拓撲結構。
共識:72.3% (722) 的 MISEV2023 調查受訪者「完全」同意, 27.0% (269) 「大致」同意第 5 節:EV 的表徵。0.3% (3) 「大致」不同意,0.4% (4) 表示他們沒有意見和/或專業知識。沒有受訪者「完全」不同意。
|
類別 |
||||
|
1. 跨膜(或 GPI 錨定)蛋白與質膜和/或內體相關 |
2. EV 中的細胞質蛋白 |
3. 非 EV 共分離結構 (NVEP) 的主要組分 |
4. 跨膜、脂質結合和可溶性蛋白與 PM/內體以外的細胞內區室相關 |
5. 與 EV 一起回收的分泌蛋白 |
|
1a: 多跨膜蛋白。 |
2b: 混雜地摻入 EV(以及可能的 NVEP)。 |
3a: 脂蛋白。 |
4a: 細胞核 |
5a: 血液來源的冠狀蛋白。 |
|
1b: 單跨膜蛋白。 |
2b: 混雜地摻入 EV(以及可能的 NVEP)。 |
3b: 蛋白質和蛋白質/核酸聚集體。 |
4b: 線粒體。 |
5b: 其他分泌蛋白。 |
|
1c: GPI- 或脂質錨定蛋白。 |
|
3c: 外泌體或超大囊泡富集組分。HSP90AA/B, TGFBI, HSPA13, LDHA/B |
4c: 分泌途徑。 |
5c: 黏附和細胞外基質蛋白。 |
|
4d: 其他 |
||||
以上內容即非經許可請勿任意複製轉發分享, 如有任何建議或詢問, 歡迎和我們聯繫
6. 關於EV 的表徵
隨著利用率和專業知識在廣泛的細胞外囊泡(EV, Extracellular Vesicle)檢測分析和儀器設備中擴展, 相關報告標準的識別也隨之增長以確保數據可靠且高重複性的說明。此處詳細列出了一份彙整的最低限度分析和儀器特定報告考量清單。
這些考量通常適用於各種實驗設計, 以下章節中列出的技術並非詳盡無遺且許多檢測技術仍在開發或正被積極研究中,然而在此所列的技術皆為市面上可得的且已有多位研究者的相關文獻支持。這些建議並非全面且鑑於實驗參數的主觀性, 可能還需要進一步的標準。
6.1 基於流式細胞術(Flow cytometry-based methods)的方法
6.1.1 基於微珠的流式細胞術 (Bead-based flow cytometry)
基於微珠的流式細胞術已被廣泛用於檢測細胞外囊泡(EV)表面蛋白。大型微珠可捕捉粒子無論其表面組成為何(例如, 無表面活性劑的醛基/硫酸鹽微珠)而抗體偶聯微珠則會捕捉暴露相應抗原的粒子。
市售的EV多重檢測試劑盒允許檢測30種或更多的表面抗原, 經捕捉後, 與微珠結合的粒子會用螢光偶聯的親和試劑(或多種混合物)做標記以進行檢測。因為訊號來自單顆粒微珠捕捉的多個粒子, 其染色強度的差異僅為半定量性, 因此, 訊號強度的差異可能意味著粒子濃度、表位密度、粒徑分布或EV亞群相對豐度有所不同。
在報告基於微珠(bead-based)的方法時, 對照組應包括做為檢測抗體的同型抗體(isotypes)或同型抗體偶聯捕獲微珠(isotype-conjugated capture beads)以及僅含檢測抗體的捕獲微珠(針對抗體包被捕獲微珠)。
可使用多種EV粒子的輸入濃度以展示訊號的滴定效應並排除非特異性結合, 以染色微珠的百分比表示並非有效的統計數據;建議報告其均一化的微珠中位數螢光強度(median fluorescence intensities); 建議從單顆微珠(singlet gated beads)中的報告資料及中位數螢光強度統計並以等效可溶性螢光分子(molecules of equivalent soluble fluorophore, MESF)(等同單顆立EV流式細胞術)表示, 以利跨儀器平台及設定資料的標準化。
若為自行製備的微珠, 應報告試劑及偶聯化學方法;若使用商業的捕獲微珠試劑則應報告目錄號及批號。其他報告參數包括:總微珠數量、樣本與微珠的孵育時間、孵育後的洗滌方法、檢測試劑染色時間及染色後的洗滌方法。
建議事項
對照組應包括做為檢測抗體的同型抗體(isotypes)或同型抗體偶聯的捕獲微球(isotype-conjugated capture beads)以及僅含檢測抗體的捕獲微球(用於抗體包被的捕獲微球)。
-
應使用多個輸入的細胞外囊泡濃度(EV concentrations)以展示訊號的滴定值(titration)。
-
如果是自行製作珠子, 應報告其試劑和偶聯化學(conjugation chemistry)。對於商用的捕獲珠子試劑則應報告目錄號和批號。
-
報告其標準化珠子的中位數螢光強度(normalised bead median fluorescence intensities)。
-
報告其數據及中位數螢光強度統計並以等效可溶性螢光分子(molecules of equivalent soluble fluorophore, MESF)(如同單顆立細胞外囊泡的流式細胞術)自單顆粒門控微球所獲得。
-
報告完整且詳細的方法論(methodology)。
6.1.2 單顆粒細胞外囊泡流式細胞術 (Single-EV flow cytometry)
流式細胞術(Flow cytometry)是一種光學技術, 在特殊情況下已被證明能檢測到約∼40奈米(nm)的囊泡(vesicles), 並且許多現代的傳統細胞儀器是利用其通過光散射(light scatter)和螢光(fluorescence), 並可檢測到約∼100奈米的囊泡。
透過數據的校準(calibration), 流式細胞術已被證明能夠表徵粒子直徑(particle diameter)、表位豐度(epitope abundance)、表位密度(epitope density)、有效折射率(effective refractive index)以及在標準化粒徑範圍內的數量濃度(number concentration)。
於2023年, 一個由三個學會組成的工作小組(EV Flow Cytometry Working Group)—涵蓋國際細胞外囊泡學會(International Society for Extracellular Vesicles)、國際細胞學促進學會(International Society for Advancement of Cytometry)及國際血栓與止血學會(International Society for Thrombosis & Haemostasis)共同發表了一份單顆粒囊泡流式細胞術的彙編(single-EV flow cytometry compendium), 全面性的探討開發單顆粒囊泡流式細胞術檢測方法的考量因素。
螢光及光散射參數的校準對於單顆粒囊泡流式細胞術結果的解讀與重現至關重要, 若使用單顆粒囊泡流式細胞術報告粒子濃度時應定義偵測極限的上下限值(LOD, limit of detection)以便使用正交技術(orthogonal techniques)重現及解讀數據。目前, 影像式細胞儀(imaging cytometers)採用的是動態觸發方法(dynamic triggering method), 這使得偵測極限的下限值(lower LOD)判斷難以界定, 因此也難以標準化。
2020年, EV流式細胞術工作組(EV Flow Cytometry Working Group)制定並發表了一套全面的實驗與報告框架(MIFlowCyt-EV)做為立場文件, MIFlowCyt-EV報告框架分為以下類別:前分析變數與實驗設計、樣本準備、分析控制、儀器校準與數據採集、細胞外囊泡(EV)表徵描述、流式細胞術(FC)數據報告及流式細胞術數據共享。該報告框架及實施MIFlowCyt-EV框架的學習資源可於EV流式細胞術工作組網站(www.evflowcytometry.org)取得。凡涉及單顆粒細胞外囊泡流式細胞術的手稿, 均須完成MIFlowCyt-EV試算表並做為補充材料附上。MIFlowCyt-EV框架適用於所有流式細胞儀, 包括傳統型、光譜型、影像型及單光子偵測型流式細胞儀。
建議事項
-
請參考 ISEV-ISAC-ISTH MIFlowCyt-EV 框架立場文件(Position Paper), 並將該報告框架做為任何使用單顆粒細胞外囊泡流式細胞術(single-EV flow cytometry)之手稿的補充材料。
-
確保體積(volume)、螢光(fluorescent)與光散射參數(light scatter parameters)的校準正確。
-
定義偵測極限的上限和下限值, 以便他人驗證您的工作。
6.2 基因蛋白標記(Genetic protein tagging)
EV的蛋白質可以透過引入一個基因構建使標記物 (例如GFP) 最終與感興趣的蛋白質或蛋白質結構域共同做轉譯從而可進行其基因標記。
所標記的蛋白質可根據其做為一般 EV 或 EV 亞型標記(第 5.7 節)的身份來選擇且目前已有多種標記物被標記過。標記蛋白質已被用於探究EV/亞型的釋放與攝取途徑並用於整體生物分布與藥物動力學研究。
重要的是, 標記物本身或標記蛋白質表達性的改變可能會影響 EV 的生物形成、裝載、釋放或功能性, 因此建議使用未標記的 EV 做為對照組以評估這些可能性。融合蛋白也可能會影響亞細胞定位或細胞功能, 應確認嵌合蛋白與野生型蛋白的定位差異。
某些標記(例如 GFP)可能會在酸性隔室中受到猝滅, 應提供其構建圖並在可能的情況下, 將質體存放於 Addgene(www.addgene.org)或其他資料庫中。
建議事項
-
請仔細考慮所選的標記蛋白及其做為細胞外囊泡(EV)或細胞外囊泡亞型(EV subtype)標記的適用性。
-
評估嵌合蛋白與野生型蛋白在細胞及細胞外囊泡(EV, extracellular vesicle)中的亞細胞定位及功能, 方法是比較其基因工程細胞與野生型細胞以及標記與未標記的細胞外囊泡。
-
報告構建的圖譜並將質粒存放於資料庫(repository)。
6.3 質譜蛋白質組學 (Mass spectrometry proteomics)
質譜法(Mass spectrometry, MS)是測量分子的質荷比(mass-to-charge ratio), 在細胞外囊泡(extracellular vesicles, EV)的研究中常用於檢測及表徵與EV相關的蛋白質, 其所涵蓋的探索性與目標性應用。
目標性分析通常在三重四極桿(triple quadrupole, QQQ)液相層析(liquid chromatography, LC)-質譜平台上進行, 而非目標性蛋白質體學則常使用飛行時間(Time-of-Flight, ToF)或Orbitrap質譜平台(Liebler & Zimmerman, 2013)進行。目標性與非目標性蛋白質體學方法在應用、優勢及樣本處理、數據獲取和分析方面都各有細微的差異。
非目標性蛋白質體學研究用於識別樣本中所有可檢測的離子, 無論是來自EV相關蛋白質或污染性的基質蛋白, 此方法是提供對樣本蛋白質組成的全面理解, 適用於生物標誌物發現等應用。針對MISEV中的EV純度(第1類、第2類)及基質污染(第3類)標記物(第5.7節)之表徵, 做目標性肽段的分析可能會更適合, 其可展示每個分析物是否有超過預先設定的檢測閾值並能定量絕對蛋白質豐度。
多重分析例如LC-MS的工作流程中, 可在有限的樣本量(如臨床試驗樣本)中提供高靈敏度分析。目標性蛋白質體學更適合定量其蛋白質的豐度變化例如疾病狀態或治療干預中的變化。結合穩定同位素標記(stable isotope labelled, SIL)肽段標準並與在匹配基質中製備的「輕」肽段校準品共同使用, 可實現對相應內源性分析物的絕對定量。
儀器設定中包括碰撞能量(collision energy)、氣體流量與溫度(gas flow and temperature)、以及毛細管電壓(capillary voltage)均依平台與分析物(analyte)而有不同, 因此應進行調適以達到最佳化並於整個專案期間保持恆定。質譜儀(MS instruments)對離子對試劑(ion-pairing reagents)、緩衝鹽(buffer salts)及去污劑(detergents)的污染極為敏感, 這會降低靈敏度與分析效能。因此, 用於目標的液相層析-質譜分析(targeted LC-MS analysis)的細胞外囊泡肽段(EV peptide)樣品應在低鹽緩衝液/質譜相容溶劑基質(low-salt buffer/MS-compatible solvent matrix)中製備, 並加入適當濃度的穩定同位素標記肽段標準品(SIL peptide standard)。
目標物分析中應包含有目標蛋白的陽性對照(positive controls)及陰性對照(negative controls), 例如來自其他物種的細胞外囊泡或不表達目標蛋白的細胞裂解液(cell lysates)。請報告目標肽段序列及肽段的選擇策略並應利用摻入適當基質中的合成輕標記與重標記肽段(synthetic light and heavy-labelled peptides)定義所響應的線性範圍及檢測與定量極限。應報告其均一化方法例如依總蛋白量、起始材料體積或蛋白質消化及注入質譜分析的顆粒數量。無論報告的目標性或非目標性蛋白質體的研究結果如何, 皆應遵循蛋白質體實驗最小資訊準則(Minimal Information About a Proteomic Experiment, MIAPE), 以促進方法學的一致性及嚴謹性/重現性。
所有樣品的製備技術應附有可重複執行的實驗描述並涵蓋每一步驟。應報告所有使用的資料軟體及版本, 以便了解資料處理方式, 亦應報告識別與定量的篩選條件、分數及信心水準以及相關時所採用的定量方法。資料及其元資料應上傳至資料庫, 以確保細胞外囊泡實驗的資料產出與報告維持嚴謹性並具有潛在的重現性。
建議事項
-
優化儀器設定並在整個專案期間保持設定不變。
-
在目標導向的液相層析-質譜(LC-MS)蛋白質分析中, 應包含正向對照(含有目標蛋白質)及負向對照組, 例如來自其他物種的細胞外囊泡(EVs)或不表達目標蛋白質的細胞裂解液。
-
將SIL肽標準品加入EV基質中以評估基質效應並展示標準品與內源性分析物之間的保留時間及定量離子與定性離子轉換比率的一致性。
-
針對目標的多反應監測(MRM)分析, 至少監測一個定量離子轉換和一個(最好是兩個)定性離子轉換。
-
使用合成的輕標記(light-labelled)和重標記(heavy-labelled)肽段加入適當的基質中以定義所響應的線性範圍以及偵測極限和定量極限。
-
報告目標肽段的序列及肽段的選擇策略以及合成肽段的同位素純度和來源。
-
樣品製備技術應包括所使用的均一化方法(normalisation approach)且應在工作流程的每個步驟中以詳細的實驗描述進行報告。
-
遵循關於蛋白質體實驗的最小資訊報告建議(Minimal Information About a Proteomic Experiment, MIAPE)。
-
將數據及元數據上傳至數據庫(data repository)
以上內容即非經許可請勿任意複製轉發分享, 如有任何建議或詢問, 歡迎和我們聯繫
6 細胞外囊泡(EV)表徵分析之技術特定的報告考量事項
隨著利用率和專業知識在廣泛的細胞外囊泡(EV, Extracellular Vesicle)檢測分析和儀器設備中擴展, 相關報告標準的識別也隨之增長以確保數據可靠且高重複性的說明。此處詳細列出了一份彙整的最低限度分析和儀器特定報告考量清單。
這些考量通常適用於各種實驗設計, 以下章節中列出的技術並非詳盡無遺且許多檢測技術仍在開發或正被積極研究中,然而在此所列的技術皆為市面上可得的且已有多位研究者的相關文獻支持。這些建議並非全面且鑑於實驗參數的主觀性, 可能還需要進一步的標準。
6.4 基於顯微鏡技術的方法
6.4.1 原子力顯微鏡(Atomic force microscopy)
原子力顯微鏡(Atomic Force Microscopy, AFM)可提供無需標記和染色的單顆粒細胞外囊泡(EVs)及共同分離的奈米粒子(nanoparticles)影像。AFM影像需要將分析物沉積於固體表面(基材, substrate), 可於樣品乾燥後進行測量或保持其浸沒於液體中如生理鹽水或細胞培養基中。
AFM形態測量(morphometry)可用於獲取EV粒徑分布及超微結構的細節並檢查污染物的存在與相對含量。此外, AFM是少數能測量單顆粒囊泡奈米機械性質(nanomechanical properties)的技術之一, 該性質被發現與EV的身份及功能性有相關聯。EV獨特的機械指紋亦可用於區分其與粒徑及形狀相似的非囊泡外泌體顆粒(Non-Vesicular Extracellular Particles, NVEPs)。
EV樣品的AFM成像最低報告要求包括對初步的樣品沉積程序、基底類型及前處理、固定方法、樣品濃度和沉積時間的詳細資訊以及任何沖洗和/或乾燥步驟的細節。應提供AFM成像模式、採集條件及探針資訊, 包括預期的探針尖端曲率半徑和彈簧常數。
如果進行定量的形態測量則應描述用於選擇測量對象的啟發式方法以及從中提取形態描述符合的程序。例如報告檢測到的顆粒高度是否大於或小於兩層脂質雙層厚度(約8奈米), 這可能有助於區分變形的EV與非EV/塌陷的EV。此外, EV機械性研究應描述為假設的接觸力學模型並理想性的提供足夠的數據, 使讀者能夠測試其替代模型。
建議事項
-
報告初步的樣品沉積程序、基板類型及前處理、固定化方法、樣品濃度及沉積次數, 並附上任何沖洗及/或乾燥步驟的詳細資料。
-
提供原子力顯微鏡(AFM)成像模式、採集條件及探針資訊, 包括預期的探針尖端曲率半徑及彈簧常數。
-
如果進行定量的形態測量, 請描述用於選擇被測量物體的啟發式方法(heuristics)以及提取形態描述符(morphological descriptors)的程序。
-
細胞外囊泡(EV)機械研究應描述假設的接觸力學模型並在理想情況下應提供足夠的數據, 使讀者能夠測試其替代模型。
6.4.2 衍射式極限型螢光顯微鏡(Diffraction-limited fluorescence microscopy)
螢光顯微鏡技術的應用範圍可以從活細胞成像到單分子定位。這些方法包括全內反射式顯微鏡(Total Internal Reflection Microscopy, TIRF-M)、共軛焦顯微鏡以及近年的光片顯微鏡, 這已被用於評估細胞與細胞外囊泡(EV)之間的相互作用, 如EV的釋放與攝取以及單顆粒EV的組成分析。
一般而言由於TIRF顯微鏡僅限於成像玻璃介面上的表面且具有高訊號的訊雜比數據使其有利於單分子偵測, 因此可能是分析EV內容物的最合適系統。共軛焦與光片顯微鏡, 尤其是最新型號的儀器可具備單分子偵測能力以進行校準及動態研究, 但其更適合用於活細胞成像實驗, 這些方法及其潛在的缺陷已被廣泛回顧。
在顯微鏡實驗中需報告顯微鏡類型、放大倍率、雷射功率及曝光時間, 因為螢光標記的樣本具有有限數量的標記分子。每個標記的樣本在光漂白(photobleaching)前只能提供有限數量的光子, 因此每次實驗必須進行優化以最大化從有限的「光子預算」(photon budget)中獲取的信息量。因此, 樣本會以最小激發量進行短時間曝光以執行活細胞實驗或以較高激發功率及較長相機曝光時間進行單分子偵測。
雖然系統校準對於定量顯微鏡實驗是必須的, 我們建議在可能的情況下, 將校準擴展至任何顯微鏡的方法中以獲得對儀器靈敏度的無偏差評估。對單顆粒螢光染料或標記蛋白分子的校準是一種成熟的方法, 能推斷存在於或內含於細胞外囊泡(EVs)上的蛋白質或RNA總數並確保即使是細胞外囊泡中僅保留少量複製的分子也能被偵測到。
應予以報告用於偵測細胞外囊泡的軟體, 包括用於設定物體強度閾值的具體參數。為此目的開發的程式碼應予以存放並向社群開放。現有的演算法是利用細胞外囊泡體積小的特性, 這些囊泡通常是繞射極限(diffraction-limited)的物體。這些演算法假設囊泡形狀與成像系統的點擴散函數(point spread function, PSF)是相同的並且在共軛焦(confocal)、全內反射螢光顯微鏡(TIRF)及光片顯微鏡(light-sheet microscopy)中可近似為高斯函數。
建議事項
-
請報告顯微鏡類型、放大倍率、雷射功率及曝光時間。
-
系統的校準(Calibration)對於顯微鏡實驗的定量是強制性的, 對於任何顯微鏡方法也建議進行以獲得對儀器靈敏度的無偏差評估。
-
應予以報告用於檢測細胞外囊泡(EVs)的軟體, 包括用於識別物體的具體參數。並且, 如適用的話應對物體強度進行閾值處理。針對這些程序所編寫的任何程式碼也應公開提供。
6.4.3 動態光散射 (Dynamic light scattering)
動態光散射(DLS), 亦稱為光子相關光譜(photon correlation spectroscopy, PCS)及準彈性光散射(quasi-elastic light scattering, QELS), 是一種能夠測定稀薄水性分散液中有足夠單分散粒子之水合動力學直徑的技術(Berne & Pecora, 1976;Hackley & Clogston, 2011;“Particle size analysis — Dynamic light scattering (DLS)” 2017;Stetefeld et al., 2016)。
DLS 可做為比色皿分析來進行或在連接流體泵(如高效液相色譜法, high-performance liquid chromatography, HPLC)時做線上的分析。水合動力學直徑是定義為一個固體球體的直徑, 其擁有與被測粒子相同的擴散係數。
DLS 是測量溶液中多個粒子的散射雷射光強度之自相關函數, 該自相關函數包含粒子擴散係數的資訊, 而擴散係數是透過布朗運動的斯托克斯-愛因斯坦理論(Stokes-Einstein theory)與水合動力學直徑相關聯。
各種演算法都可用於從測得的自相關函數推導其擴散係數, 最常見的方法為累積量分析(cumulant analysis), 該方法假設是單分散的粒徑分佈時, 但EV樣品並不符合此假設。其他方法如CONTIN演算法會試圖處理累積量分析的缺點, 但對於EV樣品的多分散粒徑分佈性而言, 從自相關函數推導擴散係數分佈會成為一個病態的數學問題。
這表示除非DLS是應用於單分散粒徑的EV分餾, 例如利用流場流動分餾法(flow field-flow fractionation)並結合線上分析的EV樣品分餾法, 否則不應使用DLS來確定EV樣品的定量性質如平均水合動力直徑(average hydrodynamic diameter)。另一方面DLS可用於定性確認EV樣品中可能存在的亞微米粒子及其可能的聚集體, 無論哪種情況都請遵循國際標準ISO 22412:2017《粒徑分析—動態光散射(Dynamic light scattering, DLS)》2017年版中關於DLS測量命名法及報告的建議。
建議事項
-
除非應用於單分散粒徑分級的細胞外囊泡(EV)樣本, 否則不應使用動態光散射(DLS)來測定EV樣本的定量性質, 例如平均水合動力直徑。
-
動態光散射(DLS)可用於定性確認EV樣品中可能存在的亞微米粒子及其可能的聚集體。
-
請遵循國際標準 ISO 22412:2017(「粒徑分析 — 動態光散射(DLS)」, 2017)中關於動態光散射(DLS)測量的命名法及報告建議。
6.4.4 電子顯微鏡 (Electron microscopy)
電子顯微鏡(Electron microscopy, EM)變體是少數能夠不論大小都可偵測細胞外囊泡(extracellular vesicles, EVs)的技術之一。然而EM的通量意味著與較小的EV相比, 較大的EV在統計上會被低估。
雖然掃描式電子顯微鏡(scanning electron microscopy, SEM)、穿透式電子顯微鏡(transmission electron microscopy, TEM)和冷凍式電子顯微鏡(cryo-electron microscopy, cryo-EM)對EV的表徵皆為高解析型方法, 但它們不一定可互換或能提供相當品質的影像。
例如cryo-EM能清楚顯示脂質雙層, 因為其可對特定體積內的所有粒子進行成像而非僅限於附著於表面(即載網)的粒子, 相較於用於固定TEM樣本的脫水條件中更能維持EV的形態且可能更具定量性。
TEM則應採用適合EV的操作流程包括使用含鈾化合物與甲基纖維素混合物進行對比的染色與包埋, 以維持脂質雙層形態。
SEM則能顯示任何大小EV的表面特徵, 但為了觀察到最小EV所需的最高放大倍率下取得的影像可能會較難分析。
在電子顯微鏡(EM)的方法中, 針對最低報告要求的標準化研究有限。
對於穿透式電子顯微鏡(TEM)則應報告三個主要參數:固定(fixation)、吸附(adsorption)和負染(negative staining)方法。固定包括:所使用的固定劑、其濃度及孵育時間。吸附包括載網材質、網格大小、薄膜類型、塗層、孵育時間及洗滌細節。負染細節應包括物質、濃度及孵育時間。應同時分享低倍與高倍放大影像, 並附上選擇的標準。
建議事項
-
穿透式電子顯微鏡(TEM)應依據適用於細胞外囊泡(EVs)的操作流程中進行, 其中包括使用含鈾化合物與甲基纖維素混合物的對比染色及包埋, 以維持雙層膜的形態。
-
任何電子顯微鏡技術使用時都應報告三大標準:固定法(fixation)、吸附法(adsorption)及負染法(negative staining methods)。
-
應同時提供高倍和低倍的放大影像用於高解析度電子顯微鏡(EV, Electron Microscopy)影像以及對樣品整體品質的評估。
6.4.5 奈米粒子追蹤分析 (Nanoparticle tracking analysis)
NTA, 也稱為單顆粒追蹤(single particle tracking)技術, 是一種在細胞外囊泡(EV)領域中廣泛使用的光學技術可用於估算粒子大小和濃度。
NTA用於測定有效折射率(effective refractive index)和表位存在性(epitope existence)的應用也已被證實。NTA通過測量粒子的擴散係數來推導其水合動力學直徑(hydrodynamic diameter), 通常實施一種算法以減少直徑分布的變異性。
需注意的是, 某些平台上使用的FTLA算法是為了更好地表示單分散混合物(monodisperse mixtures)而開發的, 而EV並非單分散混合物, 該算法可能導致假性的多峰分布(artefactual multi-modal distribution)。
目前尚無方法能夠確定或報告NTA的固定偵測極限值(LOD)並已有多項標準化研究以比較不同使用者和儀器之間的結果。由於共分離物如脂蛋白(lipoproteins)和大型蛋白複合物的計數以及直徑超過數百奈米的EV難以被量化, 使用NTA測量複雜生物液體的直徑分佈和濃度時應謹慎解讀。
NTA對粒子的檢測可通過光散射進行, 依賴於其折射率和直徑或通過螢光來進行。螢光NTA會依賴於去除未結合的標記物、染料的抗光漂白性(photobleaching resistance)以及每個粒子上存在可檢測程度的染料。
對於NTA(奈米顆粒追蹤分析)的報告應包括儀器型號、相機類型、相機設定、雷射波長、雷射功率、軟體版本、分析設定以及每幀粒子數量。如第5.2節所述, 因為NTA的統計數據容易受到偵測極限(LOD, Limit of Detection)的偏斜影響, 所以偏好報告NTA的直徑分佈而非單顆粒直徑的統計數據。若已知的話應報告用於產生直徑分佈的演算法, 因為不同演算法可能導致結果差異。
在光散射或螢光檢測模式下, 建議使用僅含緩衝液的對照組。對於螢光NTA, 應報告光散射模式下的總粒子數量及螢光模式下的標記粒子數量並說明標記移除方法及緩衝液/試劑對照組以評估標記所產生的假象。若使用樣品注射流體系統, 亦應報告其設定。
建議事項
-
應予以報告儀器型號、相機類型、相機設定、雷射波長、雷射功率、軟體版本、分析設定及每幀粒子數量。
-
報告奈米追蹤分析儀(NTA)的粒徑分佈而非單顆粒粒徑統計數據。
-
若已知的話, 請報告用於產生直徑分佈的演算法(algorithm)。
-
建議在光散射(light scatter)和螢光檢測模式(fluorescence detection modes)中均使用僅含緩衝液(buffer-only control)的對照組。
-
使用螢光奈米顆粒追蹤分析(fluorescent NTA)時, 建議報告光散射模式下的總顆粒數量、螢光模式下的標記顆粒數量, 以及標記去除方法, 並使用僅含緩衝液/試劑的對照組來評估標記假象。
-
如有使用, 請報告注射流體系統(injection fluidics)及設定。
6.4.6 單粒子干涉式反射成像感應 (Single-particle interferometric reflectance imaging sensing)
結合干涉成像/螢光成像並涉及利用親和劑(例如抗體、肽、適體)將粒子捕獲於多重陣列的微米級斑點上, 在干涉反射式成像感應器(Interference Reflectance Imaging Sensor, IRIS)模式中, 利用散射光的干涉圖樣來推導被捕獲粒子的大小與數量。
將干涉訊號轉換為名義粒徑是依賴於其折射率(Refractive Index, RI), 而折射率可能在細胞外囊泡(Extracellular Vesicles, EV)的族群中有所變異。目前的單顆粒干涉式反射成像感應器(Single Particle-IRIS, SP-IRIS)平台是假設折射率為恆定值(約1.45), 此假設可能會導致正交測量間的變異且可能會低估折射率較低的細胞外囊泡粒徑。因此建議需報告所使用的軟體版本及估計的折射率參數。
在螢光模式(fluorescence mode)中, 標記有螢光探針(fluorescent probes)的捕捉粒子會在一個或多個顏色通道中被檢測到。此模式的某些方面需要仔細考慮校準(calibrations)和對照實驗(control experiments)以獲得嚴謹的結果。例如, 對於小於繞射極限(diffraction limit)的粒子(以可見光而言, 直徑約小於∼250奈米), 需驗證所檢測事件以確認檢測到的是單顆粒粒子, 例如透過序列稀釋(dilution series)來確保每個粒子的螢光強度不會隨溶液濃度而成比例變化。
為了確認螢光特異性與細胞外囊泡(extracellular vesicles, EVs)相關, 可使用破壞囊泡的界面活性劑處理(vesicle-disrupting surfactant treatments);然而也需考慮界面活性劑可能會破壞脂蛋白粒子(lipoprotein particles)。關於螢光團(fluorophore)檢測, 以下提供報告的建議事項。
建議事項
-
報告列印於晶片上的親和試劑(affinity reagent)詳細資料。
-
報告軟體版本及估計折射率參數(estimated refractive index parameter)。
-
對於小於繞射極限(diffraction limit)的粒子, 應驗證單顆粒事件的檢測。
-
為了確認螢光特異性與細胞外囊泡(EVs, extracellular vesicles)相關, 可以使用表面活性劑來破壞囊泡(儘管它們也可能會破壞某些非囊泡外泌體顆粒(NVEPs, non-vesicular extracellular particles))。
-
對於螢光團(fluorophore)檢測, 請報告親和試劑(例如, 抗體克隆)、共軛螢光團類型、孵育濃度、光源波長、帶通濾光片截止頻率、分析軟體版本及螢光截止值, 並說明選擇這些截止值的方法。建議使用陰性對照, 如非特異性IgG捕獲點或以去除EV(EV-depleted)材料孵育的晶片, 以協助選擇這些截止值。
6.4.7 超高解析度顯微鏡(Super-resolution microscopy)
為了解析彼此距離小於光的繞射極限的螢光發射體事件,超高解析度螢光顯微鏡方法會調制光線以確保鄰近分子不會同時發射。
利用兩種主要方法可以達成比繞射極限低十倍的解析度:
(1)受激發射耗盡的顯微鏡(stimulated emission depletion, STED), 該方法利用帶有相位板的同步雙雷射系統, 空間性的調控一組螢光團的活化
(2)單分子定位顯微鏡(single molecule localization microscopy, SMLM)技術, 如(d)STORM和(f)PALM, 其時間性的調控單顆粒螢光團的隨機活化。
STED和SMLM的奈米尺度解析度非常適合用於偵測與表徵單顆粒細胞外囊泡(extracellular vesicles, EVs)及其組成, 包括EV膜層、蛋白質、DNA片段及微小核醣核酸(microRNAs, miRNAs)。透過定量分析, 這些方法會進一步被用來定義EV的大小並量化蛋白質含量以及EV中miRNA和DNA片段的定位數量。此外, STED和SMLM亦被用來做影像細胞對EV的攝取或EV群集的釋放。
超高解析度顯微鏡方法包含為樣本準備、樣本成像及數據分析量身訂做的策略。為了準備用於單分子定位顯微鏡(SMLM, Single-Molecule Localization Microscopy)及受激發射耗損顯微鏡(STED, Stimulated Emission Depletion)成像的樣本, 需使用含有適當光控螢光團(photo-controllable fluorophores)的試劑標記細胞外囊泡(EV, Extracellular Vesicle)膜層或貨物分子。
標記EV的四種典型策略包括親和標記(affinity labelling)、基因標記(genetic labelling)、共價標記(covalent labelling)以及脂溶性分子/脂質類似物的攝取(uptake of lipophilic molecules/lipid analogues)。報告的標記細節應包括標記類型、適當的試劑對照及/或參考文獻、試劑濃度、孵育時間/緩衝液, 以及去除過量螢光試劑的方法。若適用的話(例如針對分離的EV), 應報告包括蓋玻片的修飾/塗層、EV在蓋玻片上的孵育流程、固定流程, 以及親和分離的對照(例如同型抗體或非沾黏表面)。
報告的成像參數應涵蓋的主要顯微鏡元件:雷射波長(laser lines)、相機、濾光片、物鏡及其他相關光路元件。協議描述應包含詳細的成像參數, 如雷射功率、相關顯微鏡配置及成像條件(包括SMLM所用緩衝液)。多色成像報告應詳述通道間的對準情況及任何用於校正色差(chromatic aberration)的處理。
在STED(受激發射耗盡顯微鏡)中, 所得影像是由強度圖所組成, 分析通常依賴於共軛焦顯微鏡(confocal microscopy)中建立的方法;相關的處理/分析參數都應予以報告。SMLM(單分子定位顯微鏡)影像是從單分子確定的座標(即定位)重建而成, EV(細胞外囊泡)分析通常採用分割和/或聚類演算法。
為了量化SMLM中檢測到的分子密度與分子組織, 重要的是定義螢光報導分子的光物理特性(例如每個分子的平均定位數、最大暗態時間)。因此, SMLM報告應包含有影像處理參數、相關螢光報導分子的光物理特性描述以及資料分析參數/演算法的詳細資訊。新開發的分析方法都應接受評估(例如, 使用模擬或其他已驗證的方法)且自訂的程式碼皆應公開提供。
建議事項
-
報告細胞外囊泡標籤協議(EV labelling protocol)的詳細資料。
-
如適用的話, 應報告蓋玻片的修飾/塗層、在蓋玻片上孵育細胞外囊泡(EVs)的操作流程、固定程序及親和分離的對照。
-
報告主要顯微鏡組件及成像協議參數, 如雷射功率、相關顯微鏡配置及成像條件。
-
多色成像報告應詳細說明各通道之間的對齊情況及所施加的色差校正(chromatic aberration)。
-
SMLM 報告還應包括影像處理參數、相關螢光報導分子(fluorescent reporters)之光物理特性描述以及資料分析參數/演算法的詳細資訊。
-
新開發的分析方法都應該被評估且自訂編寫的程式碼都應該公開提供。
以上內容即非經許可請勿任意複製轉發分享, 如有任何建議或詢問, 歡迎和我們聯繫
6 細胞外囊泡(EV)表徵分析之技術特定的報告考量事項
隨著利用率和專業知識在廣泛的細胞外囊泡(EV, Extracellular Vesicle)檢測分析和儀器設備中擴展, 相關報告標準的識別也隨之增長以確保數據可靠且高重複性的說明。此處詳細列出了一份彙整的最低限度分析和儀器特定報告考量清單。
這些考量通常適用於各種實驗設計, 以下章節中列出的技術並非詳盡無遺且許多檢測技術仍在開發或正被積極研究中,然而在此所列的技術皆為市面上可得的且已有多位研究者的相關文獻支持。這些建議並非全面且鑑於實驗參數的主觀性, 可能還需要進一步的標準。
6.5 核酸特性分析
核酸(NAs)因其被認為具有生物標誌物(biomarker)潛力及功能性角色故是最常被檢測的細胞外囊泡(EV)成分之一。
儘管近年來有關胞外囊泡DNA在細胞間通訊及做為疾病生物標誌物的報告逐漸增多, 其中亦包括微生物感染, 但RNA的研究頻率仍遠高於DNA。一些早期的細胞外囊泡研究報告指出DNA存在於細胞外囊泡腔內, 而近期的部分研究則建議DNA主要會與細胞外囊泡表面相關聯。
這些看似矛盾的發現可能是由於缺乏標準化方法來保護細胞外囊泡表面DNA在細胞外囊泡分離與表徵分析過程中免於被消化的原因, 而細胞外囊泡DNA的主要類型是單股DNA(ssDNA)還是雙股DNA(dsDNA)仍存在爭議。
RNA 研究面臨的挑戰包括輸入量、均一化和靈敏度, 其同樣適用於細胞外囊泡(EV)DNA研究中, 大多數對 EV-RNA 的表徵描述會涉及以下一項或多項包含:檢測、鑑定、定量、定位(在 EV 內部或外部)及富集(包裝)。低輸入量的 RNA 定序(RNA-Seq)和定量聚合酶鏈反應(qPCR)常用於鑑定 EV 製備中的特定序列。
國際細胞外囊泡學會(ISEV)先前已針對 EV-RNA 研究的各個方面提供指導, 涵蓋從樣本收集到生物資訊分析, 美國國立衛生研究院(NIH)細胞外 RNA 通訊聯盟(ERCC)亦有相關指導。
無論RNA表徵方法為何, RNA純化及檢測前準備過程皆可能會引入偏差因素。一些RNA的純化方法主要是分離較長的RNA(>200核苷酸(nt)), 而其他方法則因設計偏向於濃縮短RNA。對於RNA-Seq, 文庫製備方法可能會選擇特定尺寸範圍內的RNA或插入片段。逆轉錄(Reverse transcription)協議亦可能會選擇特定RNA例如帶有polyA尾的轉錄本。
基於平鋪探針(Tiled probe-based)影像技術對較長RNA的檢測, 針對較短或降解的轉錄本其靈敏度會降低。以接頭連接(Adapter ligation)為基礎的小RNA文庫製備方法, 針對miRNA優化, 同時也會富集含有5′-磷酸(5′-phosphates)及3′-羥基(3′-OH)的其他RNA, 而具有不同末端化學結構的RNA則會被低估。
高度結構化的RNA, 如全長tRNA, 除非使用耐高溫酶, 否則逆轉錄效率不佳。因此, 準確解讀及報告結果會依賴於充分理解並詳細報告技術以評估其偏差性。
由於其能夠檢測及測量少量核酸, 逆轉錄即時定量聚合酶鏈反應(reverse transcription real-time quantitative PCR, qPCR)在細胞外囊泡(EV)領域被廣泛應用。我們建議qPCR實驗盡可能遵循定量即時PCR實驗發表最低資訊標準(Minimum Information for Publication of Quantitative Real-Time PCR Experiments, MIQE)指引以及2017年國際細胞外囊泡學會(ISEV)立場文件中的ISEV EV RNA檢查清單。
在分享qPCR結果時, 除了報告均一化或處理後的數據外, 還應提供原始的定量週期數(cycle of quantitation, cq)值, 以便讀者評估目標RNA的豐度及檢測的可靠性。雖然cq值會受多種變數影響, 單獨看可能資訊有限, 但其通常與豐度會呈現相關性, 尤其是在液態樣本中, 且可報告所輸入的樣本體積時更是如此。
或許並非所有MIQE的原則皆廣泛適用於細胞外樣本。例如當樣本中僅含有極微量的載體特異性RNA時, 要確保每個樣本輸入的RNA量相同可能並不總是可行。鑑於細胞外樣本的液態特性, 有些研究者偏好以樣本輸入體積進行均一化。均一化的策略會大幅影響結果的解讀且應列於報告中。
數位PCR(digital PCR), 包括液滴數位PCR(droplet digital PCR, ddPCR), 可提供絕對定量值且已被證實相較於傳統qPCR能提升EV-RNA檢測的重現性與準確性, 絕對定量值亦可能避免均一化相關的問題。
建議事項
-
對於基於qPCR(定量聚合酶鏈反應)的分析, 請報告逆轉錄接頭(reverse transcription adapters)或引物(primers)的序列以及擴增步驟中使用的引物和探針(probes)(如適用)、實驗設計包括生物學和技術複本、精確的循環條件以及數據納入和排除標準。
-
對於RNA-Seq, 請報告所有核酸片段化(nucleic acid fragmentation)、逆轉錄(reverse transcription)、接頭(adapters)及接頭連接(接合或無接合)(adapter attachment (ligation or ligation-free))、擴增(amplification)與多重化(multiplexing)以及純化或尺寸選擇(clean-up or size selection)的所有細節。
-
對於定序數據分析, 報告預處理、序列比對、重疊註解與資料庫品質、定量及均一化/差異表達分析。
6.6 蛋白質與非蛋白質標記的細胞外囊泡(EVs)
大多數細胞外囊泡(EV)標記試劑包含其螢光基團(fluorescence moieties), 但其他檢測模式亦可使用且應採用類似的對照措施。
由於細胞外囊泡體積小因而其載貨容量有限, 蛋白質及非蛋白質標記物的檢測是困難且易受到標記過程中未結合試劑或分離方法中共分離物的干擾。未結合標記物需移除的程度會隨著技術靈敏度的提升而增加。例如, 對於能檢測少於10分子試劑的技術, 如超高解析顯微鏡(super-resolution microscopy)、表面增強干涉式反射成像系統(SP-IRIS)及單顆粒粒細胞外囊泡流式細胞術(single EV flow cytometry), 未結合染料的存在可能會輕易導致假陽性事件。
脂質染劑(Lipid dyes)常用於結合或插入至細胞外囊泡(EV)膜中。脂質專一性(Lipid-specificity)並不能保證細胞外囊泡專一性(EV-specificity), 因為非囊泡外泌體顆粒(NVEPs)如脂蛋白(lipoproteins)可能會被共同分離並被染色且某些試劑也可能會標記蛋白質, 因此建議補充使用細胞外囊泡蛋白標記(EV protein marker)。脂質標記(Lipid labels)亦可能自我聚集(self-aggregate)且對不同膜組成的細胞外囊泡親和力(affinity)會有所差異。
標記EV表面的蛋白質反應性染料(Protein-reactive dyes)也可能標記游離蛋白質及含蛋白質的非囊泡外泌體顆粒(protein-containing NVEPs)。若EV分離方法無法完全去除非EV成分, 則應認識並/或評估此可能性。如前所述, 可使用脂質標記物(lipid marker)來補充蛋白質標記。當蛋白質偽影(protein artefacts)成為可能出現時, 可使用低濃度去污劑(detergent)來評估EV膜的易變性(lability)及相關訊號的減少。
對於抗體, 製造商匹配的同型對照(isotype controls)可用於與特異性抗體相同的濃度做使用, 這是支持特異性的一種方法。陰性EV對照(negative EV controls), 例如來自不表達抗體表位(epitope)的細胞, 也是有用的對照之一。
在需要於染色後進行純化的檢測中, 應使用程序控制以證明純化前後的細胞外囊泡(EV)族群一致性、純化程序未引入人工產物, 且染料應已被去除。例如:
-
在標籤去除方法(例如, SEC分離)之前和之後, 使用試劑對照分析緩衝液以評估游離/聚集標籤的去除情況。
-
分析未染色的細胞外囊泡(EVs)在標籤去除方法(label depletion method)前後的情況以證明該方法不會改變或選擇性富集細胞外囊泡群體。
-
分析標記去除後的試劑染色樣本並與未染色結果(上述)進行比較以評估可能的染料誘導變化。染色尤其可能會顯著增加小型細胞外囊泡(small EVs)的直徑或密度。
建議事項
-
使用緩衝液/僅標記對照以識別由未結合標記引起的假陽性偽影。然而, 僅標記偽影並非唯一可能的標記偽影。
-
對於抗體, 製造商匹配的同型對照(isotype controls)可用於與特異性抗體相同的濃度。
-
抗體用於評估結合特異性。也可使用陰性EV的對照(缺乏抗體表位)。
-
請注意, EV 蛋白標記方法可能同時標記游離蛋白質及含蛋白質的非囊泡外泌體顆粒(NVEPs), 而脂質染劑可能標記含脂質的非囊泡外泌體顆粒(NVEPs)。請確保 EV 分離方法適用於後續分析。如果 EV 分離方法無法完全去除非 EV 成分, 則應認識並/或評估此可能性。
-
為了識別非EV標記偽影(non-EV labelling artefacts)的貢獻, 建議同時使用蛋白質(protein)和脂質(lipid)標記。
-
在染色後需要進行純化的檢測中, 應使用程序控制以證明純化前後的細胞外囊泡(EV)群體一致性、純化程序未引入假象且多餘的染料已被去除。
6.7 拉曼光譜分析(Raman spectroscopy)
拉曼光譜學(Raman spectroscopy, RS)是一種無標記的分析光學技術, 能夠基於樣品在窄線寬雷射照射下產生的非彈性散射光子以定性及定量解析樣品小體積的化學組成。
拉曼光譜本質上是雷射光束焦點內被測小體積樣品的化學指紋。RS能實現化學特異性、非破壞性探測, 樣品前處理需求極少或無, 且對被測樣品的水含量相對不敏感。為克服RS訊號微弱的問題, 其中一種策略是使用表面增強拉曼散射(surface-enhanced Raman scattering, SERS), 這是RS的一種奈米等離子體輔助放大衍生技術。此方法是利用金屬奈米結構將拉曼散射訊號放大數個數量級。
自發拉曼與表面增強拉曼兩種方法均已證明在基礎研究及轉譯性細胞外囊泡(extracellular vesicles, EV)分析中具有實用價值。
拉曼光譜中的裝置間(inter-device)及裝置內(intra-device)變異性可能由多種原因引起, 包括雷射變異以及各光學元件(包括探測器)對不同光能量的非均勻響應(稱為光譜響應 spectral response), 因此拉曼系統應進行仔細校正。現代商用拉曼系統具備自動校正程序, 但舊型及實驗室自製系統則無此功能, 這會進而增加重現性的問題。測量的多個面向應列在報告中, 包括雷射波長與功率、校正程序、主要光學元件的製造商及型號、物鏡的數值孔徑(numerical aperture)及放大倍率(如適用的話)、探頭類型及規格(通常適用於非顯微鏡設置與測量)以及雷射光斑的物理尺寸。
光譜採集參數亦應說明, 例如每個樣本或採樣點所收集的光譜總數、每次光譜訊號收集時間(亦稱積分時間或採集時間 integration/acquisition time)以及掃描時掃描區域或體積的尺寸(例如:100 × 100區域, 步進大小400奈米, 總掃描區域40微米 × 40微米), 最後是建議報告中要包含所有與樣品製備相關的參數。
由於細胞外囊泡(EV, extracellular vesicles)樣本通常懸浮於含有不同濃度溶解化合物的水溶液中因而具有不同的滲透壓, 故需考慮並報告EV的配方及其測量時是懸浮樣態或乾燥樣態。例如, EV可利用表面增強拉曼探針(SERS nanoprobes)於懸浮樣態中測量或乾燥於石英玻璃載片上進行拉曼光譜採集。
目前尚不清楚濕態測量與乾態測量相較是否具有優勢, 因此只要詳細說明EV樣本製備步驟, 兩種方法皆被視為可行。
除了儀器和樣本的考量外, 資料分析和統計程序也會影響RS研究的終點和結論。所有資料的分析軟體及其版本皆應列在報告中。
若使用自訂程式的套件和演算法, 建議將程式碼存放於線上資料庫以確保透明度與可重複使用性; 於取得資料後(且在後續分析之前)欲相互比較的光譜應使用相同的資料處理參數進行後處理。例如, 若實施基線校正(baseline correction)和/或背景扣除(background subtraction), 所有的相關參數應對所有光譜保持一致。所有後續的光譜分析及進一步的統計檢定(例如, 多變量分析、多機器學習、統計假設檢定)皆應完整報告, 並公開相關資料。
建議事項
-
報告所有儀器和測量參數。
-
報告樣品製備/應用參數, 包括緩衝液組成及濕測/乾測。
-
報告數據分析軟體及版本。將任何自訂的程式套件及演算法的程式碼存放於線上數據庫以確保透明度及可重複使用性。
-
報告下游頻譜分析及進一步的統計檢測。
6.8 電阻脈衝感應(Resistive pulse sensing)
電阻脈衝感應(Resistive pulse sensing, RPS)是一種非光學技術, 其利用庫爾特原理(Coulter principle)來測定粒子的濃度與直徑並在某些平台上可同時測量界達電位(zeta potential)。
目前RPS的實作包括預先校準的固定孔徑微流體卡匣格式以及未校準的可伸縮薄膜孔徑, 兩者的偵測下限均可達約∼50奈米(nm)直徑且能測量粒徑高達數微米的粒子。利用RPS測量複雜生物液體中細胞外囊泡(extracellular vesicles, EVs)的直徑分布與濃度時應謹慎解讀, 因為共同分離物(co-isolates), 如脂蛋白(lipoproteins)和大型蛋白質複合體, 也會被計數且無法與EVs區分。然而, RPS測量結果與穿透式電子顯微鏡(transmission electron microscopy, TEM)的數據具有高度一致性。
在報告電阻脈衝感應(RPS)數據時, 建議應報告儀器型號、孔徑大小、校準顆粒直徑及來源以及軟體版本。
對於可伸縮的薄膜孔徑, 應共享所施加的電壓、施加的拉伸量及優化設定的程序。
對於微流控電阻脈衝感應, 應考慮並報告適當的稀釋緩衝液以降低水的表面張力。
如第5.2節所述, 對於細胞外囊泡(EV)的數據, 較佳的做法是報告RPS的直徑分佈而非單顆粒直徑統計數據, 因為RPS統計數據容易受到偵測極限(LOD)的下限偏斜影響。建議同時包含僅含緩衝液的對照組以識別背景訊號以及在相同濃度下使用去污劑裂解的樣本以確定標記事件。
由於RPS技術容易因較大顆粒而堵塞, 可能會需要使用離心或過濾等前分析步驟以去除較大顆粒。由於這些方法可能改變所分析的EV族群並影響與正交方法的比較, 任何前分析程序應明確說明。
建議事項
-
報告在RPS(電阻脈衝感應)之前所施行的任何前分析程序。
-
對於微流控電阻脈衝感應(microfluidic RPS), 應考慮並報告適當的稀釋緩衝液(dilution buffer)。
-
包含僅含緩衝液的對照組以及以與未處理樣品相同濃度進行的洗滌劑裂解樣品(detergent-lysed samples)。
-
報告所有儀器及軟體細節。
-
報告RPS直徑分佈, 而非單顆粒直徑統計數據。
6.9 西方墨點法(Western blotting)
西方墨點法(Western blotting)是一種常用於檢測含有細胞外囊泡(EV, extracellular vesicle)製備物中蛋白質的方法。蛋白質首先通過凝膠電泳分離, 然後轉移到膜上, 並使用親和試劑(通常是抗體)進行探測。
其輸入量通常會根據EV製備物的某些指標(總蛋白質量、顆粒數量)或EV來源的某些指標(生物液體體積、培養細胞數量)進行均一化:前者允許比較相似EV群體之間的EV貨物量而後者則可能評估來源系統中EV的產生與攝取平衡的整體差異。
對於細胞培養來源的EV, 應將細胞裂解液(以指定蛋白質量或細胞等效量計)與EV樣品同時加載於同一凝膠中以評估EV相較於細胞生成的富集或耗竭情況。然而此比較僅能輕易的進行於來自細胞培養條件培養基的EV分析, 因為對於其他EV來源(例如生物樣本), 來源細胞無法輕易識別或回收。
在可能的情況下, 應在實驗樣本旁邊加入已知的抗原陽性和陰性對照樣本。若聲稱蛋白質是存在於EVs上或內部時 亦應包含用於評估樣本製備純度的對照組, 詳見第5.7節。
應報告其抗體資訊(特異性、克隆、來源、標記濃度、孵育時間)、樣本變性條件、還原劑的存在與性質、轉移方法、膜類型、緩衝液以及成像設備和參數。為了有足夠的透明性, 建議至少以補充資料形式來提供未裁剪的西方墨點(Western blot)影像(包括對照和分子量標準)。
建議事項
-
請提供蛋白質富集(protein enrichment)及定量(quantification)的詳細資料。
-
在可能的情況下, 提供包含抗原陽性(antigen-positive)和抗原陰性(antigen-negative)的對照組。
-
如果聲稱蛋白質與細胞外囊泡(EV, Extracellular Vesicle)有相關, 需包含細胞外囊泡製備純度的測量指標。
-
報告所有輸入的均一化(input normalization)、凝膠電泳(gel electrophoresis)、轉移方法(transfer methodology)、探針(probing)及成像/分析(imaging/analysis)的詳細資訊。這些內容包括但不限於抗體資訊(antibody information)、樣品變性及還原條件(sample denaturing and reducing conditions)、轉移方法(transfer methodology)、膜類型(membrane type)、緩衝液(buffers)以及成像設備與參數(imaging equipment and parameters)。
-
請提供所有西方墨點法(Western blots)的未裁剪影像(例如, 若發表於期刊中, 可做為補充資料提供)。
共識:70.6%(705位)參與MISEV2023調查的受訪者「完全」同意第6節:針對EV(細胞外囊泡)特徵描述的技術特定報告考量, 27.5%(274位)「大部分」同意。0.4%(4位)「大部分」不同意, 1.5%(15位)表示他們沒有意見和/或專業知識。沒有受訪者「完全」
以上內容即非經許可請勿任意複製轉發分享, 如有任何建議或詢問, 歡迎和我們聯繫
 外泌體尺寸排阻色譜分離管柱qEV columns (SEC) [GMP-Ready]
外泌體尺寸排阻色譜分離管柱qEV columns (SEC) [GMP-Ready] 外泌體尺寸排阻色譜後濃縮試劑盒Nanotrap Extracellular Vesicles Particles
外泌體尺寸排阻色譜後濃縮試劑盒Nanotrap Extracellular Vesicles Particles 外泌體RNA提取試劑組Norgen Exosomal RNA Isolation Kit
外泌體RNA提取試劑組Norgen Exosomal RNA Isolation Kit 一次性血細胞計數盤Disposable Hemocytometer
一次性血細胞計數盤Disposable Hemocytometer 奈米磁珠分選細胞組Magnetic microbeads Cell Seperation Products
奈米磁珠分選細胞組Magnetic microbeads Cell Seperation Products 工業級切向流過濾系統qEV TFF
工業級切向流過濾系統qEV TFF 次世代外泌體自動餾份收集機AFC-V2
次世代外泌體自動餾份收集機AFC-V2 大規模外泌體尺寸排阻分離自動色譜系統qEV Zenco
大規模外泌體尺寸排阻分離自動色譜系統qEV Zenco 標準型大規模外泌體尺寸排阻分離自動色譜系統qEV Zenco Standard
標準型大規模外泌體尺寸排阻分離自動色譜系統qEV Zenco Standard cGMP大規模外泌體尺寸排阻分離自動色譜系統qEV Zenco Pro
cGMP大規模外泌體尺寸排阻分離自動色譜系統qEV Zenco Pro 工業級cGMP大規模外泌體尺寸排阻分離自動色譜系統qEV BigZenco
工業級cGMP大規模外泌體尺寸排阻分離自動色譜系統qEV BigZenco 高通量外泌體自動機械人純化系統qEV Dxter
高通量外泌體自動機械人純化系統qEV Dxter 高穩定細胞電穿孔系統 Extransction Electroporation System
高穩定細胞電穿孔系統 Extransction Electroporation System 組織均質與單細胞懸浮液製備儀DSC Single Cell Suspension Dissociator
組織均質與單細胞懸浮液製備儀DSC Single Cell Suspension Dissociator 酸鹼計、電導度計、溶氧度計、離子濃度計、濁度計、色度計(電化學與水質檢測指示儀)Electrochemical & Water Quality Analyzer
酸鹼計、電導度計、溶氧度計、離子濃度計、濁度計、色度計(電化學與水質檢測指示儀)Electrochemical & Water Quality Analyzer 精密微量電子天平Analytical Balance
精密微量電子天平Analytical Balance 智能攪拌機S-100 Intelligent Stirrer
智能攪拌機S-100 Intelligent Stirrer 單顆粒奈米粒徑/數量濃度/Zeta膜電位分析儀Pulsoid Nanoparticle Analyzer (NPS)
單顆粒奈米粒徑/數量濃度/Zeta膜電位分析儀Pulsoid Nanoparticle Analyzer (NPS) 單顆粒奈米粒徑/數量濃度/Zeta膜電位分析儀Exoid Nanoparticle Analyzer (TRPS)
單顆粒奈米粒徑/數量濃度/Zeta膜電位分析儀Exoid Nanoparticle Analyzer (TRPS) 超微量全波長分光光度計Nabi UV/Vis Nano Spectrophotometer
超微量全波長分光光度計Nabi UV/Vis Nano Spectrophotometer 微孔盤全波長多功能分光光度計Mobi UV/Vis Microplate Spectrophotometer
微孔盤全波長多功能分光光度計Mobi UV/Vis Microplate Spectrophotometer 超靈敏單管冷光計Lumi Single Tube Luminometer
超靈敏單管冷光計Lumi Single Tube Luminometer 自動/高通量細胞計數儀EVE PLUS / EVE HT Automatic Cell Counter
自動/高通量細胞計數儀EVE PLUS / EVE HT Automatic Cell Counter 自動螢光細胞計數儀ADAM-MC / ADAM-MC2 / ADAM-CellT Automated Fluorescence Cell Counter
自動螢光細胞計數儀ADAM-MC / ADAM-MC2 / ADAM-CellT Automated Fluorescence Cell Counter 全類型分光光度計Spectrophotometer
全類型分光光度計Spectrophotometer 一鍵式蛋白轉移系統EZ BLOT One-Touch Transfer System
一鍵式蛋白轉移系統EZ BLOT One-Touch Transfer System 接觸式化學冷光成像儀e-BLOT WB/SDS-Pages Touch Imager
接觸式化學冷光成像儀e-BLOT WB/SDS-Pages Touch Imager 超恆定二氧化碳細胞培養箱D181 Incubator
超恆定二氧化碳細胞培養箱D181 Incubator 高速(冷凍)微量離心機High-Speed (Refrigerated) MicroCentrifuge
高速(冷凍)微量離心機High-Speed (Refrigerated) MicroCentrifuge