
日本PMDA發佈: 外泌體Exosome(細胞外囊泡, EVs)臨床產品之品管和安全性指南白皮書
日本PMDA(Pharmaceuticals and Medical Devices Agency,醫藥品醫療機器總合機構)是日本的藥品與醫療器械監管機構, 成立於2004年4月1日,是一個獨立的行政法人機構。其核心使命是通過確保藥品和醫療器械的安全性、有效性和品質來保護公眾健康, 與日本厚生勞動省為共同合作運作。
2024年8月, PMDA官方的Science Board發佈了近40頁的白皮書, 近似於一步步的拆解外泌體Exosome/細胞外囊泡EVs的治療產品從細胞庫、GMP流程、品管實施、非臨床(研究級)到FIH的全部細節, 諾曼達科技為您解析如下:
日本PMDA《基於細胞外囊泡的治療產品之品質與安全性考量》指南深度解讀
發布單位: 日本醫藥品醫療機器總合機構 (PMDA) 科學委員會
發布日期: 2024年8月7日
文件來源: Quality and Safety Considerations for Therapeutic Products Based on Extracellular Vesicles
1. 指南概述
本指南由日本PMDA的科學委員會發布,旨在全面評估基於細胞外囊泡(Extracellular Vesicles, EVs)的治療產品在開發、製造、品質控制及安全性評估方面所面臨的挑戰與考量。EVs作為體內天然的分子傳遞系統,因其靶向能力、生物相容性及低免疫原性等優點,在藥物開發和藥物遞送系統(DDS)領域展現出巨大潛力。然而,其臨床應用仍面臨諸多挑戰。本指南匯總了日本專家的最新研究討論,為EVs的臨床應用提供了科學和監管層面的指導,期望能促進新型EV療法的發展。
2. 核心概念與分類
指南首先對EVs進行了定義和分類,強調了其作為細胞間通訊工具的重要性。
EVs是由細胞釋放、具有脂質雙層膜的囊泡,其內部包裹著蛋白質、核酸(如mRNA、miRNA)等功能性分子。它們通過體液運輸,將這些分子遞送至靶細胞,從而改變接收細胞的表型和基因表達。
根據國際細胞外囊泡學會(ISEV)的建議,指南將EVs分為兩大類:
-
天然EVs (Native EVs): 來自未經基因改造或經過基因改造但無轉基因產物的細胞。這類EVs利用其內源性物質發揮治療作用,例如,來自間質幹細胞(MSC)的EVs具有抗炎和組織修復的潛力。
-
工程化EVs (Engineered EVs): 通過基因工程改造生產細胞,或在EVs純化後對其表面進行修飾、裝載特定藥物(如siRNA、小分子藥物)而製成。這類EVs主要作為藥物遞送系統(DDS),旨在提高藥物的靶向性和穩定性。
3. 品質與製造考量 (CMC)
指南的核心部分詳細闡述了EVs產品在製造與品質控制方面的關鍵考量,強調了標準化和一致性的重要性。
3.1. 起始材料:細胞來源的選擇
EVs的特性高度依賴於其來源細胞。
3.2. 細胞庫的建立與鑑定
為了確保生產的一致性,指南強調必須建立主細胞庫(MCB)和工作細胞庫(WCB)。
3.3. EVs的表徵與分析
由於EVs的高度異質性,單一的分析方法不足以全面評估其品質。
3.4 EVs的製造方式
3-4-1. 細胞來源選擇與細胞庫建立(Cell source & Cell Bank)
細胞是 EVs 品質的根源,而EVs 的 cargo、活性成分以及功能特性會高度依賴細胞的狀態、來源及純度, 因此 PMDA 指南強調必須:
- 建立 MCB/WCB 或 DCB(視自體/異體/細胞系而定)
- 確認細胞身分(identity)、表面標誌、基因穩定性
- 針對 immortalized 或 genetically modified cell lines 做基因載體(vector)序列確認與複本數測定
3-4-2. 製造培養條件(Culture Conditions)
PMDA 指南指出EVs分泌與 EVs 的組成會受到多種因素影響,如:
- 培養基組成(FBS、hPL、細胞激素等)
- 細胞密度
- 氧氣、CO₂、溫度條件
- 細胞凋亡比例(apoptotic EVs 具有完全不同的生物功能)
FBS 與 hPL 的重點:
- FBS 不建議用於 GMP(除非有特殊用途), 因為這會有引入 bovine EVs、病毒/內毒素的風險
- hPL 為人源替代品,但仍需驗證感染風險與殘留量
3-4-3. 細胞狀態與製程一致性(In-process controls)
正式純化前必須確認:
- 接種後細胞狀態:型態、活率、凋亡比例
- 培養上清中的 EVs 數量是否符合預期
- EVs 內部預期的活性成分是否達標
這些是確保後續純化的 EVs 批次一致性的重要步驟。
3.5 EVs的純化方法(EV Purification Methods)
PMDA 明確指出目前無「黃金標準」,須視產品性質組合多種方法。
純化的瓶頸與原則
- EVs 與病毒(特別是 enveloped virus)大小相近 → 不易分開
- EVs 與蛋白複合體、脂蛋白、聚合物也常共沈或共流出
不同大小的 EVs(sEV、m/lEV)有不同活性,需視目標選擇方法
PMDA 指南解析主流的純化方法和建議:
[1]. 超高速離心(Ultracentrifugation, UC)
常見於研究階段,但:
- 難以放大量
- 容易 co-isolate free proteins
- 無法作為商業化製程
通常 UC 只適合作為初步濃縮,並需要搭配其他的方法。
[2]. PEG 或其他聚合物沉澱(Precipitation by PEG 等)
缺點明確:
- 大量蛋白聚集物與雜質
- 需建立殘留測定方法(因 PEG 可能留在製劑中)
[3]. 親和色譜(Affinity Chromatography)
可依 EV 表面蛋白進行純化,例如:
- CD9/CD63/CD81 抗體
- 或針對 engineered EV 上的特定蛋白⁽如 GE11, RVG peptide⁾
但需注意:
- EV markers ≠ 所有 EV 都有
- 得確認「被純化到的子群」是否仍具活性
[4]. 粒徑為基礎的方式(Size-based methods)
- SEC (Size Exclusion Chromatography)
- Ultrafiltration (MWCO membranes)
優點:可 scale-up大體積進行,產物有均一性
缺點:可能混入有蛋白聚集體、脂蛋白或與 EV 相近大小的物質,需要進行殘留測定
[5]. 離子交換色譜(Ion-exchange chromatography)
利用 EV 表面電荷分離(如 anion exchange)
- 可達較高純度
- 已有研究成功 scale-up大體積生產
[6]. 多方法整合(Combination methods)
PMDA 強調需要的流程為:
濃縮(Concentration)→ 初濾去除雜質 → 粒徑/電荷/親和色譜的組合
此可達到商業化與 GMP 等級的一致性。
3.5 EVs 特異性的品質表徵方式(EV-specific Quality Characterization)
是這份指南中最重要的核心內容之一
3-5-1. 為何 EVs的QC 很困難?
- EVs 是有 高度異質性(heterogeneous)的粒子族群
- 不可能像蛋白藥(monoclonal antibody)那樣去定義單一分子
- 每顆 EVs 的 cargo 都可能不同
- EVs 與 lipoprotein、virus、protein complex 大小重疊
因此 QC 需以多種正交方法(orthogonal)來驗證。
3-5-2. EVs 組成分析(Composition analysis)
需要分析的成分有:
- 蛋白質(Western blot, ELISA, Mass spec)
- 脂質
- RNA(miRNA, mRNA)
- 醣鏈(glycan)
- 表面 tetraspanins(CD9、CD63、CD81)
- 內涵物(TSG101、Alix)
注意事項: MISEV 建議至少 3 個 EV markers,但 PMDA 認為不必完全遵照研究領域標準而是需依產品特性決定。
3-5-3. 物理化學特性(Physicochemical properties)
[1]. 粒徑分析(Size distribution)
建議使用 單顆粒分析方法(single particle analysis):
- NTA (Nanoparticle Tracking Analysis)
- TRPS (Tunable resistive pulse sensing)
- FCS (Fluorescence correlation spectroscopy)
- EM (TEM/SEM)
- AFM (Atomic force microscopy)
注意:不推薦用DLS, 因為 EVs 屬於是 polydisperse這會導致DLS 容易失真。
[2]. 粒子數(Particle count)
主要使用:
- NTA
- TRPS
[3]. 表面電位(Zeta potential)
- ELS (Electrophoretic light scattering), 但PMDA 也提到新的「單顆粒 zeta potential」技術正在發展中。
3-5-4. 生物活性(Biological Activity / Potency Assay)
這是最關鍵的 QC 要求。
Potency assay 用於:
- 「證明」EV 具預期的生物機制(MoA)
- 「量化」批次間活性
- 作為臨床給藥單位(dose unit)基準(如:per particle, per µg protein)
常見 potency assays:
- miRNA/protein 功能測試
- 抗發炎作用(IL-1β/ TNF-α 抑制)
- 免疫調節(T cell suppression、NK activation)
- 血管新生(endothelial tube formation)
- 纖維化抑制
- 細胞遷移/修復
PMDA 特別強調:EVs 的作用機制通常無法完全明確,因此必須使用多個 orthogonal potency assay
3-5-5. EVs 製劑中的雜質(Impurities)— 特別重要
PMDA 將 EV 雜質分為:
- 非預期 EVs(未含活性成分)
- 製造過程產生的 altered EVs / degraded EVs
- 來自細胞本身:mitochondria、Golgi vesicles
- 來自培養液:FBS / hPL EVs, serum protein complex
- 感染源:virus / mycoplasma
- 實驗器材的微粒(plastic debris)
因此 QC 必須包含「雜質的定性與定量」。
4. 非臨床與臨床安全性的考量
指南對EVs產品的安全性評估提出了系統性要求。
非臨床研究:
- 藥物動力學 (PK):由於EVs的異質性,傳統PK研究難以進行。指南建議使用上述標記方法追蹤EVs在體內的分佈、吸收和清除。
- 毒理學研究:需評估EVs的靶向毒性(On-target toxicity)和脫靶毒性(Off-target toxicity)。特別是對於工程化EVs,需同時評估EV載體本身和其裝載藥物的毒性。
- 免疫原性:需評估EVs引發的不良免疫反應,特別是來自非人源細胞(如植物、牛乳)的EVs。
臨床研究:
- 首次人體試驗 (FIH):起始劑量的確定是一個挑戰。指南建議基於非臨床研究的毒理數據(如NOAEL)和效價測定結果,謹慎設計起始劑量。
免疫原性風險:在臨床試驗中需密切監測免疫反應,特別是重複給藥時可能產生的抗體。
5. 圖表解讀
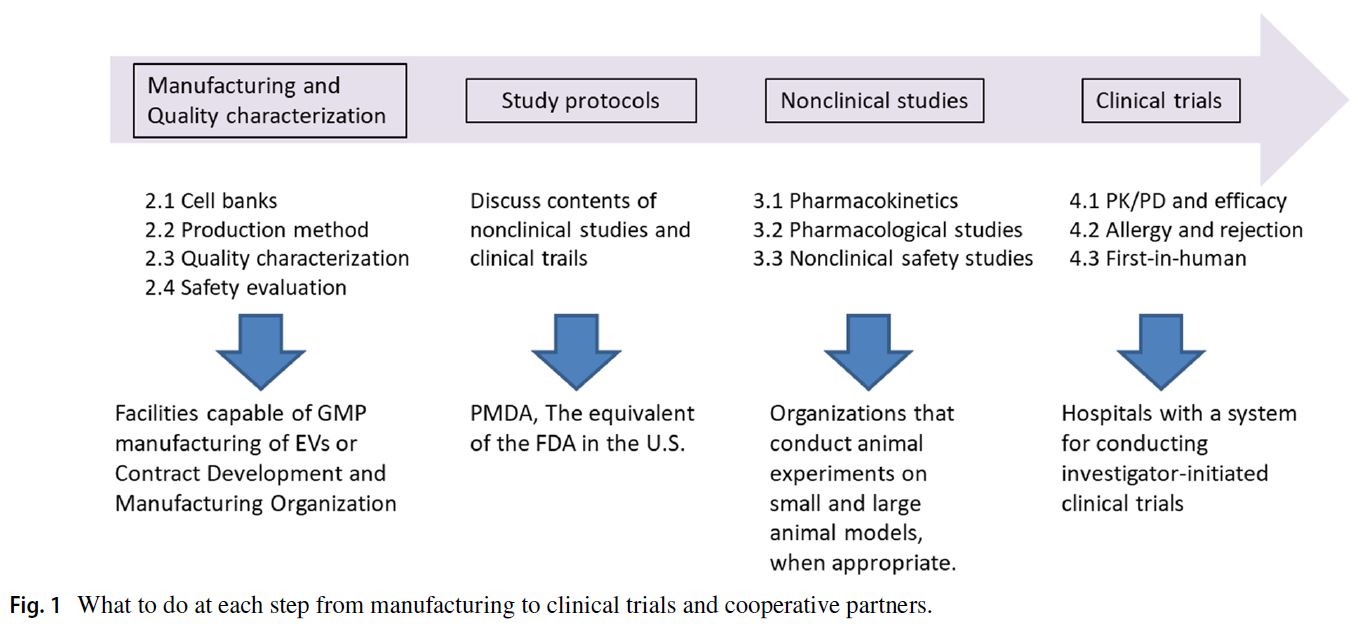
Figure 1: EVs 治療產品開發的路線圖(Roadmap)
此圖其實是一個宏觀的開發框架圖,在 PMDA 指南中指出「開發 EV 藥物一定要做哪些事」的流程。
內容分為 五大區段:
1. 製程方法開發(Manufacturing Method Development)
製程建立、細胞來源選擇、培養條件、純化方法、製程一致性等內容
核心工作內容
- 選擇並建立 EVs 製造用細胞(Primary cells、MSC、iPS/ES-derived cells、immortalized lines)
- 建立 細胞庫(Cell Bank:MCB/WCB/DCB)
- 建立標準化的培養流程
- 控制培養環境避免污染(病毒、細菌、內毒素等)
- 選擇並建立合適的 EVs 純化技術 (例如 SEC、ion-exchange、ultrafiltration、affinity、組合式流程)
合作夥伴
- 細胞治療專家
- 生產工廠(GMP manufacturing)
- 生物加工工程師(bioprocess engineers
2. 品質表徵分析(Quality Characterization)
核心工作內容
確立 EVs 的 關鍵品質屬性(CQAs)
- 組成:蛋白、RNA、脂質、糖鏈
- EVs markers:CD9、CD63、CD81、TSG101、Alix…
- 粒徑分布、粒子數量、Zeta potential界達電位
- 生物功能(potency assay)
- 建立一致的測定方法(如NTA、DLS、TRPS、Western、Mass spec…)
合作夥伴
- 分析化學家
- EVs 研究學者
- 品質管制(QC)單位
3. 非臨床研究(Nonclinical Studies)
核心工作內容
生物分佈(biodistribution/PK)
- EVs 標定(PKH, DiR, RI, USPIO…)
- 評估器官分布、滯留時間、清除途徑
藥效(Pharmacology)
- 使用細胞、動物模型確認治療機制與作用
毒理(Toxicology)
- 重複給藥毒性
- 安全性指標
- 外來物污染評估(virus, mycoplasma)
合作夥伴
- 動物實驗研究人員
- 藥理毒理專家
- 成像與 EVs 標定技術專家
4. 臨床開發(Clinical Trials)
核心工作內容
- FIH(First-in-Human)劑量設定(基於 MABEL 或 NOAEL)
- 安全性監測
- 與製造批次一致性比對
- 評估臨床療效
- 長期追蹤 EVs 生成細胞的安全性(若為 engineered EVs)
合作夥伴
- 臨床醫師
- 統計與臨床研究團隊
- 法規專家(Regulatory Affairs)
5. 整體的跨領域合作網絡(Collaborative Partners)
Figure 1 強調了整個開發流程的高度跨領域性,包括:
- 細胞/幹細胞研究者
- EVs 生物學家
- 製程工程師
- 分析與品控(QC/QA)
- 動物實驗與成像專家
- 臨床研究者
- 法規人員(PMDA/FDA/etc.)
核心訊息總結:
- EVs 產品開發不是單一步驟,而是一條完整管線:細胞建立 → 製程開發 → EVs 純化 → 品質確認 → 非臨床 → 臨床。
- 每一階段都必須建立明確的標準化流程(SOP/GMP)
- 品質特性(CQAs)與 potency assay 是最核心的開發難點
- 病毒安全(Viral safety)是 EV 特有且重大的風險點
- 整個流程高度依賴跨領域整合與合作
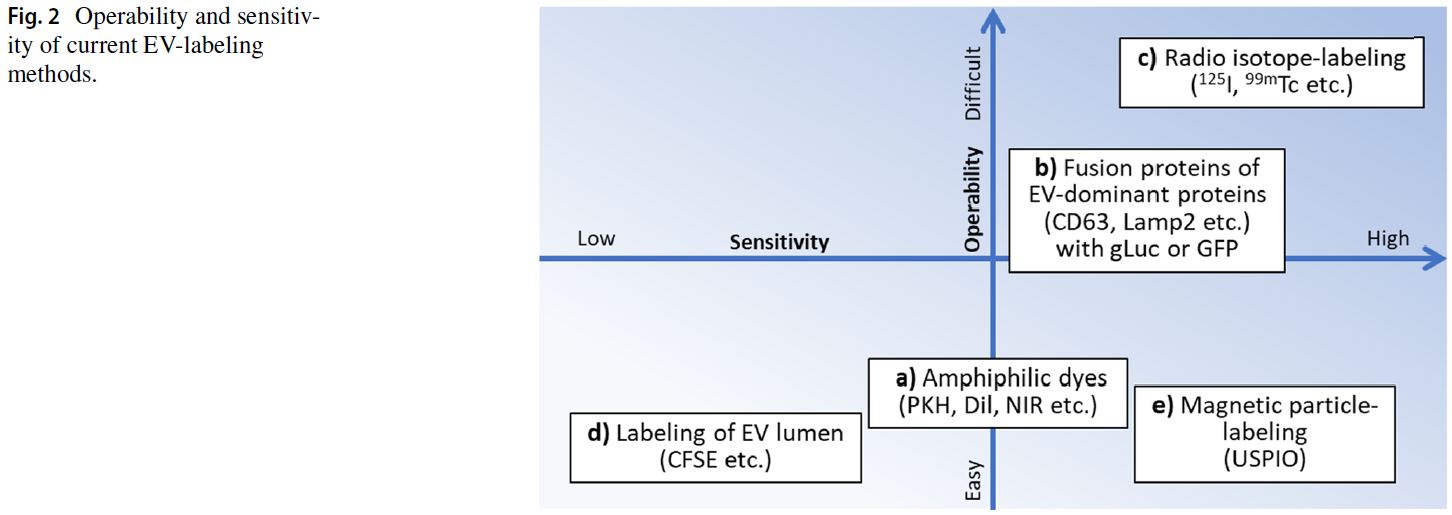
Figure 2: EVs標記方法的可操作性與靈敏度
此圖為研究人員在進行EVs體內分佈(生物分佈)研究時,選擇標記方法提供了清晰的決策依據。圖表將不同的標記技術根據「操作難易度」和「偵測靈敏度」進行了分類。
- 左下象限 (操作簡單,靈敏度低):如CFSE等染料,適合初步或定性的示蹤研究。
- 右下象限 (操作簡單,靈敏度較高):如兩親性染料 (PKH, DiI) 和 磁性粒子 (USPIO),在操作便利性和偵測效果之間取得了較好的平衡,是目前應用較廣泛的方法。
- 右上象限 (操作困難,靈敏度高):如放射性同位素標記 (¹²⁵I) 和 融合蛋白技術 (CD63-GFP),雖然操作複雜、成本高,但能提供最精確的定量數據,適用於後期的藥物動力學研究。
Table 1 討論了幾種主要細胞來源的優缺點:
|
細胞來源 |
優點 |
缺點與風險 |
|
自體/同種異體初代細胞 |
- 安全性高- 更接近天然生理狀態 |
- 難以標準化- 產量有限- 批次間差異大 |
|
iPS/ES細胞衍生細胞 |
- 可大規模生產- 可建立標準化細胞庫 |
- 存在致瘤性風險- 表型不穩定 |
|
永生化細胞株 |
- 可大規模、標準化生產- 技術成熟 |
- 存在致癌基因污染的理論風險- 長期培養下表型可能不穩定 |
Table 2 詳細列出了對各級細胞庫進行鑑定的要求,總結如下:
|
測試項目 |
供體細胞庫 (DCB) |
主細胞庫 (MCB) |
工作細胞庫 (WCB) |
體外培養極限細胞 (LIVCA) |
|
表徵鑑定 |
✓ |
✓ |
✓ |
✓ |
|
純度測試 |
✓ |
✓ |
✓ |
✓ |
|
無菌測試 |
✓ |
✓ |
✓ |
✓ |
|
支原體測試 |
✓ |
✓ |
✓ |
✓ |
|
病毒測試 |
✓ |
✓ |
✓ |
✓ |
|
儲存穩定性 |
✓ |
✓ |
(適用時) |
- |
|
基因表現構建體分析 |
(適用時) |
(適用時) |
(適用時) |
(適用時) |
註:LIVCA(Limit of In Vitro Cell Age)指用於生產的細胞達到其體外培養壽命極限的狀態,對其測試是為了確保整個生產週期內的穩定性。
Table 3 總結了用於EVs表徵的多種分析方法,涵蓋了生物物理、生物化學及功能性分析。
|
分析類別 |
具體項目 |
常用技術方法 |
|
生物物理特性 |
粒徑分佈、濃度、型態、Zeta電位 |
NTA, TRPS, DLS, 電子顯微鏡 (TEM, SEM), AFM |
|
生物化學特性 |
總蛋白、總核酸、總脂質、EV標記物 (CD9, CD63, CD81)、非EV標記物、活性成分 |
BCA/Bradford Assay, RiboGreen Assay, 質譜, 西方墨點法, 流式細胞術, ELISA |
|
生物學活性 |
效價測定 (Potency Assay) |
細胞活性測試 (增殖、遷移、凋亡), 基因/蛋白表達分析, 體內動物模型 |
6. 結論
日本PMDA的這份指南為全球EVs治療產品的開發者提供了寶貴的參考框架。它不僅系統性地梳理了從生產製造到臨床應用的全鏈路考量,還重點強調了當前技術的局限性和未來需要努力的方向。核心觀點可以總結為:
- 標準化是關鍵:無論是細胞來源、生產工藝還是品質檢測,都必須建立嚴格的標準化流程,以確保產品的一致性和可比性。
- 全面性的表徵:必須採用多種互補的分析方法,從物理、化學和生物學多個維度對EVs進行全面表徵。
- 風險控制:應基於科學和風險評估的原則,對潛在的安全性風險(如致瘤性、免疫原性)進行充分識別和控制。
總體而言,本指南的發布標誌著EVs治療正從基礎研究向規範化的藥物開發邁進,為該領域的健康發展奠定了重要的監管科學基礎。
以上內容即非經許可請勿任意複製轉發分享, 如有任何建議或詢問, 歡迎和我們聯繫
 外泌體尺寸排阻色譜分離管柱qEV columns (SEC) [GMP-Ready]
外泌體尺寸排阻色譜分離管柱qEV columns (SEC) [GMP-Ready] 外泌體尺寸排阻色譜後濃縮試劑盒Nanotrap Extracellular Vesicles Particles
外泌體尺寸排阻色譜後濃縮試劑盒Nanotrap Extracellular Vesicles Particles 外泌體RNA提取試劑組Norgen Exosomal RNA Isolation Kit
外泌體RNA提取試劑組Norgen Exosomal RNA Isolation Kit 一次性血細胞計數盤Disposable Hemocytometer
一次性血細胞計數盤Disposable Hemocytometer 奈米磁珠分選細胞組Magnetic microbeads Cell Seperation Products
奈米磁珠分選細胞組Magnetic microbeads Cell Seperation Products 工業級切向流過濾系統qEV TFF
工業級切向流過濾系統qEV TFF 次世代外泌體自動餾份收集機AFC-V2
次世代外泌體自動餾份收集機AFC-V2 大規模外泌體尺寸排阻分離自動色譜系統qEV Zenco
大規模外泌體尺寸排阻分離自動色譜系統qEV Zenco 標準型大規模外泌體尺寸排阻分離自動色譜系統qEV Zenco Standard
標準型大規模外泌體尺寸排阻分離自動色譜系統qEV Zenco Standard cGMP大規模外泌體尺寸排阻分離自動色譜系統qEV Zenco Pro
cGMP大規模外泌體尺寸排阻分離自動色譜系統qEV Zenco Pro 工業級cGMP大規模外泌體尺寸排阻分離自動色譜系統qEV BigZenco
工業級cGMP大規模外泌體尺寸排阻分離自動色譜系統qEV BigZenco 高通量外泌體自動機械人純化系統qEV Dxter
高通量外泌體自動機械人純化系統qEV Dxter 高穩定細胞電穿孔系統 Extransction Electroporation System
高穩定細胞電穿孔系統 Extransction Electroporation System 組織均質與單細胞懸浮液製備儀DSC Single Cell Suspension Dissociator
組織均質與單細胞懸浮液製備儀DSC Single Cell Suspension Dissociator 酸鹼計、電導度計、溶氧度計、離子濃度計、濁度計、色度計(電化學與水質檢測指示儀)Electrochemical & Water Quality Analyzer
酸鹼計、電導度計、溶氧度計、離子濃度計、濁度計、色度計(電化學與水質檢測指示儀)Electrochemical & Water Quality Analyzer 精密微量電子天平Analytical Balance
精密微量電子天平Analytical Balance 智能攪拌機S-100 Intelligent Stirrer
智能攪拌機S-100 Intelligent Stirrer 單顆粒奈米粒徑/數量濃度/Zeta膜電位分析儀Pulsoid Nanoparticle Analyzer (NPS)
單顆粒奈米粒徑/數量濃度/Zeta膜電位分析儀Pulsoid Nanoparticle Analyzer (NPS) 單顆粒奈米粒徑/數量濃度/Zeta膜電位分析儀Exoid Nanoparticle Analyzer (TRPS)
單顆粒奈米粒徑/數量濃度/Zeta膜電位分析儀Exoid Nanoparticle Analyzer (TRPS) 超微量全波長分光光度計Nabi UV/Vis Nano Spectrophotometer
超微量全波長分光光度計Nabi UV/Vis Nano Spectrophotometer 微孔盤全波長多功能分光光度計Mobi UV/Vis Microplate Spectrophotometer
微孔盤全波長多功能分光光度計Mobi UV/Vis Microplate Spectrophotometer 超靈敏單管冷光計Lumi Single Tube Luminometer
超靈敏單管冷光計Lumi Single Tube Luminometer 自動/高通量細胞計數儀EVE PLUS / EVE HT Automatic Cell Counter
自動/高通量細胞計數儀EVE PLUS / EVE HT Automatic Cell Counter 自動螢光細胞計數儀ADAM-MC / ADAM-MC2 / ADAM-CellT Automated Fluorescence Cell Counter
自動螢光細胞計數儀ADAM-MC / ADAM-MC2 / ADAM-CellT Automated Fluorescence Cell Counter 全類型分光光度計Spectrophotometer
全類型分光光度計Spectrophotometer 一鍵式蛋白轉移系統EZ BLOT One-Touch Transfer System
一鍵式蛋白轉移系統EZ BLOT One-Touch Transfer System 接觸式化學冷光成像儀e-BLOT WB/SDS-Pages Touch Imager
接觸式化學冷光成像儀e-BLOT WB/SDS-Pages Touch Imager 超恆定二氧化碳細胞培養箱D181 Incubator
超恆定二氧化碳細胞培養箱D181 Incubator 高速(冷凍)微量離心機High-Speed (Refrigerated) MicroCentrifuge
高速(冷凍)微量離心機High-Speed (Refrigerated) MicroCentrifuge